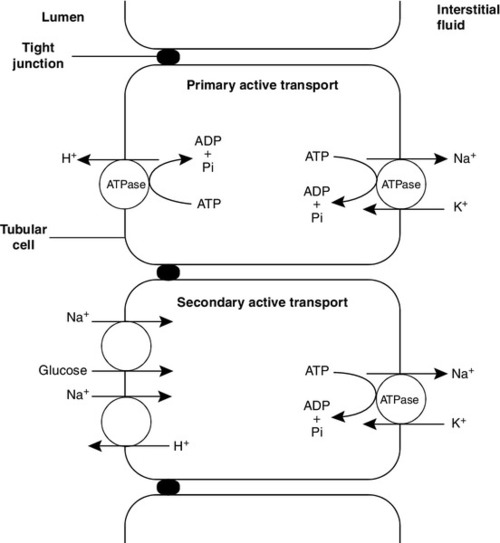
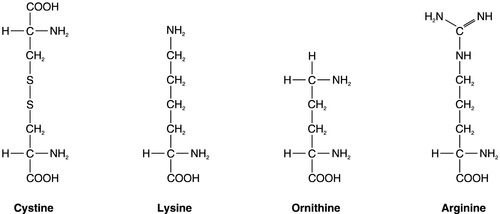
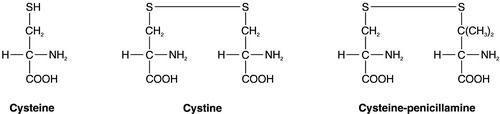
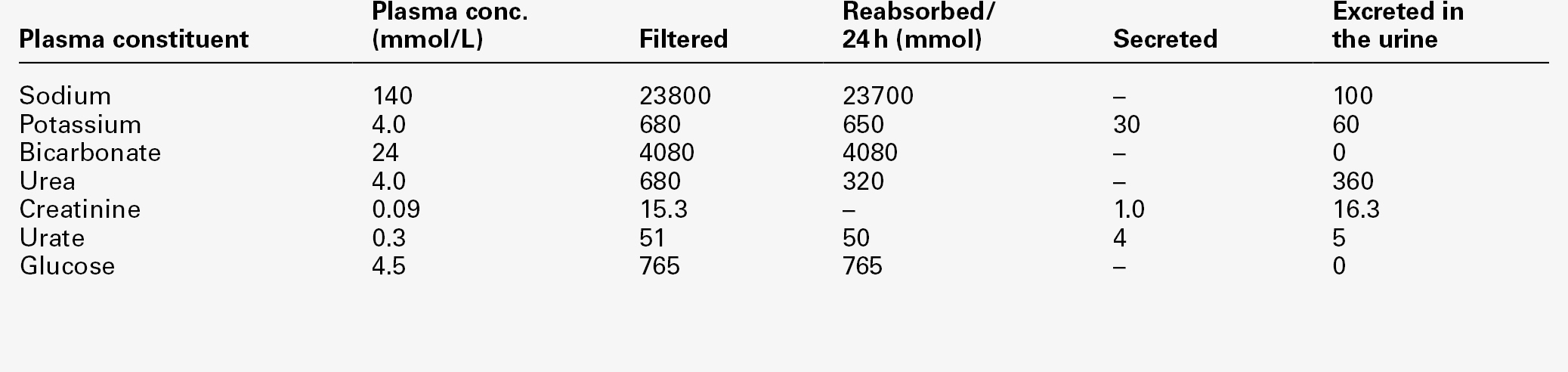
CHAPTER 9 David Makanjuola; Marta Lapsley CHAPTER OUTLINE Isolated abnormalities of tubular function Generalized tubular defects (Fanconi syndrome) Investigation of stone formers Most patients with renal disease have some element of renal tubular involvement, but the other manifestations of the disease tend to be clinically more obvious and important. However, in a small number of patients, the clinical picture results primarily from a disorder of renal tubular function. These disorders can be inherited or acquired, and can affect tubular handling of a limited number of specific substances or encompass more generalized defects. Renal tubular defects are conveniently considered with renal stone formation, since calculi sometimes form as a result of one of these conditions. Hereditary renal tubular disease includes certain developmental disorders of the tubules, for example polycystic renal disease and medullary cystic disease. While these can result in disorders of renal function, including renal tubular function, they will not be considered in detail here. Renal tubular physiology will be discussed briefly, followed by a discussion of some well-recognized functional disorders of the renal tubules. Renal function is discussed in detail in Chapter 7, but in essence the process involves filtration at the glomeruli, followed by modification of this glomerular filtrate by both tubular reabsorption and tubular secretion. Since 170 L of filtrate are formed each 24 h, but only about one-hundredth this amount of urine is produced, reabsorption is quantitatively the more significant (Table 9.1). This is largely an active, energy-requiring process and explains why the kidneys account for some 6–8% of the resting oxygen consumption of the body, while representing < 1% of body mass. Some of the mechanisms by which active transport in the renal tubules occurs are shown in Figure 9.1. The control of renal tubular handling of certain substances is covered in detail in other chapters, for example sodium and water in Chapter 4. Only the renal tubular handling of substances that are important in disorders of renal tubular function will be considered further here. FIGURE 9.1 Active transport mechanisms in the renal tubule. Glucose is absorbed with sodium ions in the early part of the proximal tubules, in a secondary active transport process. Glucose and sodium bind to a common carrier protein (SGLT 2, see later) in the luminal membrane and sodium moves down its electrochemical gradient, carrying glucose into the cell. Na+,K+-ATPase in the non-luminal (basolateral) membrane of the tubular cells pumps the sodium ions out into the interstitial fluid, while glucose is transported in the same direction by the glucose transporter GLUT 2. Amino acids are also reabsorbed in the early part of the proximal renal tubules, again by a secondary active transport system linked to sodium reabsorption. There appear to be separate cotransporter proteins for certain groups of amino acids, although some of these probably have overlapping specificities. The process is driven by the Na+,K+-ATPase in the basolateral membrane pumping sodium out of the cell, with amino acids leaving by passive or facilitated diffusion. Phosphate reabsorption in the renal tubules is influenced by the dietary intake of phosphate, certain hormones and a variety of other factors, and these are described in Chapter 6. In summary, about 90% of the inorganic phosphate in the plasma is freely filtered at the glomeruli, and then about 75% is reabsorbed in the proximal tubules. A small, variable amount is also absorbed in the distal tubules, but overall reabsorption is incomplete and up to 40 mmol/24 h appears in normal adult urine. The rate-limiting step in reabsorption appears to be a secondary active transport system linked to sodium reabsorption, with a phosphate/sodium cotransporter located in the luminal membrane of the tubular cells. As described in Chapter 5, the renal tubular secretion of hydrogen ions is linked to the net effective reabsorption of bicarbonate (see Figs 5.2 and 5.3). Around 4000 mmol of bicarbonate is filtered every 24 h, but normal urine contains virtually no bicarbonate so the tubules must secrete 4000 mmol of hydrogen ions to achieve this. Although this mechanism prevents the loss of alkali into the urine, it does not result in the net excretion of acid. The tubules must also secrete the hydrogen ions produced each day by normal metabolism (see Chapter 5), a further 40–80 mmol/24 h. There are two distinct mechanisms by which hydrogen ions are secreted into the tubular lumen (Fig. 9.1). A secondary active transport system linked to sodium operates in the epithelial cells of the early tubular segments, so that the Na+,K+-ATPase on the basolateral membrane produces an electrochemical gradient for sodium to enter the cell from the luminal surface, but in contrast to glucose and amino acids, a hydrogen ion is simultaneously secreted into the lumen. Although a very high hydrogen ion gradient cannot be achieved, this mechanism is responsible for the bulk of hydrogen ion secretion, so that most bicarbonate reabsorption occurs in the proximal tubules. The sodium–bicarbonate cotransporter, located on the basolateral membrane of these tubular cells, mediates the transport of the generated bicarbonate into the systemic circulation. There may be other hydrogen ion secretory mechanisms in the proximal tubules, but they do not appear to be quantitatively important. In the late tubular segments (the late distal tubules and the collecting ducts), a completely different mechanism for hydrogen ion secretion exists. This is relatively independent of tubular sodium content and occurs through primary active transport. The intercalated cells (I cells) in this part of the nephron have a hydrogen ion-transporting ATPase on their luminal surfaces and, although this accounts for < 5% of the total hydrogen ions secreted, it is important because it can generate a hydrogen ion gradient of almost 1000 to 1. It is this that is responsible for the final acidification of urine and dictates the minimum achievable urinary pH of about 4.5 (or maximum hydrogen ion concentration of ~ 32 μmol/L). A chloride–bicarbonate exchanger at the basolateral membrane of the intercalated tubular cells is responsible for transporting the bicarbonate generated during this process to the systemic circulation. Glucose is freely filtered at the glomeruli, but is normally then reabsorbed in the proximal tubules so that it is undetectable in urine. If plasma glucose concentrations rise to greater than ~ 10 mmol/L or if the glomerular filtration rate increases (as in pregnancy), then the capacity of the proximal tubules to reabsorb filtered glucose is exceeded and glycosuria occurs. Generalized defects in renal tubular function may also result in glycosuria (see later), but a small group of people appear to have an isolated defect of tubular glucose reabsorption. Patients with this condition excrete a variable amount of glucose in their urine at normal plasma glucose concentrations. Other aspects of carbohydrate metabolism are not affected: glucose tolerance and plasma insulin concentrations are normal. The condition is inherited in an autosomal recessive manner, and two major phenotypes have been identified (types A and B), based on the exact changes in the kinetics of glucose reabsorption. Hereditary renal glycosuria is rare, and is generally recognized to be a benign condition with no clinical sequelae. The reabsorption of glucose in the proximal renal tubules is similar to the absorption of glucose in the intestine. There is a Na+,D-glucose cotransporter on the luminal cell wall to transport glucose into the cell, with a facilitated glucose transporter (GLUT 2) on the basolateral membrane to enable the glucose to exit. The intestinal form of the Na+,D-glucose cotransporter (SGLT 1) and its gene have been well characterized, but, while the corresponding renal cotransporter (SGLT 2) is known to differ from SGLT 1, its identity in humans is less well established. Nevertheless, it is assumed that mutations in the gene for SGLT 2 cause hereditary renal glycosuria by interfering with the uptake of glucose in the proximal tubules. There is a corresponding condition affecting SGLT 1 in the gut. This cotransporter is involved in both glucose and galactose absorption, and the classic presentation is with life-threatening diarrhoea in early infancy due to glucose and galactose malabsorption (familial glucose–galactose malabsorption). There is often an associated mild renal glycosuria, although in hereditary renal glycosuria there is no corresponding effect on the gut. Mutations affecting GLUT 2, which facilitates transport of glucose, galactose and fructose across the basolateral membrane, are another rare cause of renal glycosuria (Fanconi–Bickel syndrome). Amino acids are normally freely filtered at the glomeruli and then almost entirely reabsorbed in the proximal convoluted tubules. There is a maximal capacity to each reabsorptive mechanism and, in most patients with amino aciduria, some extrarenal disorder leads to accumulation of amino acid(s) in the plasma, that exceeds the reabsorptive capacity of the tubules, with consequent ‘overflow’ amino aciduria. However, as with glycosuria, generalized defects in renal tubular function can result in amino aciduria, and there are also some isolated defects in the reabsorption of particular groups of amino acids. Cystinuria is the classic example of an amino aciduria due to a defect in renal tubular function, in that the amino aciduria occurs at normal or even low plasma concentrations of the amino acids involved. In most patients with cystinuria, there is renal loss not only of cystine, but also of the dibasic amino acids ornithine, arginine and lysine. There is also an associated failure of intestinal absorption of the same amino acids. Inspection of their molecular structures (Fig. 9.2) shows that each has two amino groups separated by 5–7 bonds, which suggests that malfunction of a single membrane carrier protein might explain the disorder. However, the true explanation is not this simple, since the clearance of cystine may exceed the creatinine clearance, suggesting secretion of cystine into the tubules and, furthermore, since dibasic amino aciduria (e.g. lysinuric protein intolerance) or cystinuria can each occasionally occur alone. FIGURE 9.2 Chemical structures of the dibasic amino acids involved in cystinuria. The only known clinical manifestation of cystinuria is recurrent urinary tract stone formation, the name ‘cystine’ coming from the original (erroneous) assumption that the source of these stones was the bladder. Cystine stones form readily in acidic urine. They are yellow-brown in colour and are radio-opaque because of their sulphur content, although they are less radio-dense than calcium-containing stones. There may also be some calcium deposition if there is infection secondary to the calculi. They tend to occur as staghorn or multiple recurrent stones and often require some form of surgical intervention or lithotripsy (fragmentation of stones by external shock waves). Patients with cystinuria also have a higher incidence of calcium oxalate stones than the general population. All stone formers should, therefore, be screened for cystinuria, preferably by formal amino acid measurement in the urine, as qualitative screening tests are not sufficiently sensitive. The intestinal defect appears to cause no clinical problems. Cystinuria occurs with equal frequency in both sexes, although males tend to be more severely affected. It may present at any time from the first year of life up to the ninth decade, with a peak incidence in the second and third decades. The prevalence of cystinuria varies between racial groups and according to whether the figures are taken from neonatal amino acid screening programmes or from known cystinuric stone formers (immaturity of the renal tubules in the first few months of life may lead to some heterozygous infants having urinary cystine outputs in the homozygous cystinuria range, leading to misclassification). However, the worldwide prevalence is estimated to be 1 in 7000, making it a relatively common inherited metabolic disorder. The mode of inheritance is autosomal recessive, although in some families it appears to be incompletely recessive, with heterozygotes excreting more urinary cystine, ornithine, arginine and lysine than normal, although less than in the homozygous state. In the past, this has led to attempts to classify cystinuria into type I (homozygous for the fully recessive form) and types II and III (variants of the incompletely recessive form), based on combinations of the relative amounts of the amino acids excreted in the urine and/or the intestinal uptake of cystine and dibasic amino acids. The discovery of the genes involved in the recessive and the incompletely recessive forms of cystinuria has made this classification clearer, although the situation is not yet completely resolved. The gene involved in type I cystinuria codes for a protein known as rBAT (a loose acronym for ‘related to the b0,+ amino acid transporter’, with the term b0,+ signifying broad specificity for neutral (0) and dibasic (+) amino acids). rBAT is a membrane glycoprotein that is one of the activating components of a heteromultimeric transporter for cystine and dibasic amino acids. The gene for rBAT (SLC3A1) is located at 2p16.3-p21, and, to date, more than 120 different mutations have been identified in it in patients with type I cystinuria. A second cystinuria locus, accounting for non-type I forms of cystinuria, has been identified by linkage analysis at chromosome 19q13.1–13.2. This gene (SLC7A9) encodes a protein that acts as the amino acid transporting subunit of the membrane complex. More than 95 gene mutations have been identified. Most, but not all cases of cystinuria can be explained by mutations in SLC3A1 and SLC7A9. Healthy individuals excrete < 10 μmol cystine/mmol of creatinine (< 130 μmol/24 h). Patients with homozygous cystinuria usually excrete > 1700 μmol/24 h and may excrete up to 5000 μmol/24 h. Heterozygotes may have an entirely normal amino acid excretion pattern in the urine or may excrete up to 1700 μmol cystine/24 h, in which case they may produce stones. Medical treatment of cystinuria begins with maintenance of a high fluid intake throughout both day and night and alkalinization of the urine, both being aimed at decreasing the chance of precipitation of cystine in the renal tract. The solubility of cystine is pH dependent with a solubility limit of ~ 700 μmol/L at pH 7, rising to ~ 1500 μmol/L at pH 7.5. Regular monitoring is required to ensure that cystine remains well below its solubility limit throughout the day and, critically, the night. If these measures fail, then it may be possible to convert the cystine to a more soluble compound, most commonly by the use of D-penicillamine. This can form the mixed disulphide cysteine-penicillamine (Fig. 9.3), which is significantly more soluble than cystine. The aim is to keep the daily excretion of free cystine below 2000 μmol. Unfortunately, D-penicillamine frequently causes an allergic reaction and can also cause nephrotic syndrome and pancytopenia, so careful monitoring is essential. Second generation chelating agents include tiopronin (α-mercaptopropionylglycine) and captopril, although studies on the efficacy of the latter have been inconclusive. Small stones may dissolve with careful medical management, although frequent surgical intervention is required for some patients. FIGURE 9.3 Chemical structures of cysteine, cystine and cysteine-penicillamine. Occasionally, in spite of both medical and surgical treatment for stone formation, cystinuria causes sufficient renal damage to result in established renal failure. In this case, renal transplantation may be effective, since the donor kidney should not be affected by the amino acid transport defect and the patient should, therefore, remain disease free. A second example of an amino aciduria due to a true defect in renal tubular function, rather than an ‘overflow’ effect, is found in Hartnup disorder. This is named after the family in which it was first described and is again a defect of both renal and intestinal amino acid transport. The constant feature is a failure to reabsorb the neutral amino acids (Box 9.1) in the renal tubules, with their consequent appearance in the urine. The failure of reabsorption is not absolute, as renal clearances of the affected amino acids are generally lower than the creatinine clearance. Most affected individuals also have increased amounts of indoles (e.g. indican) in the urine, which originate from the bacterial breakdown of unabsorbed tryptophan in the gut. Rarely, the renal or intestinal lesions may occur alone. The original description of Hartnup disease included a pellagra-like skin rash, transient cerebellar ataxia and constant renal amino aciduria. Some affected individuals have also shown psychotic behaviour, while others have learning disabilities. The pellagra-like rash and its response to nicotinamide suggest that the clinical features of the disease may be due to failure to absorb tryptophan in the intestine and reabsorb it in the renal tubules, leading to a deficiency of nicotinamide. However, investigation of siblings of individuals with Hartnup disease and the results of neonatal urine amino acid screening programmes both suggest that the typical amino aciduria (Hartnup disorder) may exist without the features of Hartnup disease. It would appear that Hartnup disorder is an autosomal recessive inherited condition, but that this is not expressed clinically without the presence of certain other genetic or environmental influences, such as poor nutrition. The causative gene, SLC6A19, is located on chromosome 5p15.33 and codes for a sodium-dependent neutral amino acid transporter. A further example of a specific abnormality in renal tubular reabsorption causing a distinct pattern of amino/imino aciduria occurs in familial renal iminoglycinuria. The primary genetic defect is a mutation in either the SLC36A2 gene, which codes for a high affinity glycine, proline and hydroxyproline transporter, or the SLC36A1 gene, which codes for a low affinity transporter. However, the phenotype is variable as it may be modified by mutations in at least three other genes. Some reabsorption of these amino acids can still occur via the transport system that is not affected in any individual patient. The condition, which is benign, is autosomal recessive, although some heterozygotes are ‘incomplete’ and have hyperglycinuria. In some but not all homozygotes, impaired intestinal transport of proline can be demonstrated. Neonatal screening for amino acidurias suggests that familial renal iminoglycinuria occurs in 1 in 15 000 live births in a Caucasian population. However, it should be appreciated that in healthy neonates, the renal tubular reabsorption of proline, hydroxyproline and glycine is less efficient than in adults; imino aciduria normally disappears by three months and hyperglycinuria by six months of age.
Renal tubular disorders and renal stone disease
INTRODUCTION
RENAL TUBULAR DISORDERS
Introduction
Physiology
Isolated abnormalities of tubular function
Glycosuria
Hereditary renal glycosuria
Amino acidurias
Cystinuria
Hartnup disorder
Familial renal iminoglycinuria
Dent disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


