RED BLOOD CELL DISORDERS
ANEMIAS
 Definition
Definition
Anemia is a reduction in Hb leading to decrease in oxygen supply to peripheral tissues. Normal hemoglobin (Hb) range is established by population studies, but range should be adjusted for different age groups, especially for children, and levels are lower in women and in African Americans. There is some debate whether people of older ages have physiologically lower Hg levels. Most likely lower values reflect underlying pathology. Hb values are more accurate than hematocrit (Hct) values, because Hb is measured directly by automated analyzers, whereas the Hct is a calculated value.
 Diagnosis
Diagnosis
 There are many ways to classify anemias, but the differential diagnosis of anemia can be narrowed by using the RBC size, as reflected in the MCV and the reticulocyte count. See Figure 9-1.
There are many ways to classify anemias, but the differential diagnosis of anemia can be narrowed by using the RBC size, as reflected in the MCV and the reticulocyte count. See Figure 9-1.
 In addition, insight into mechanism and etiology complements the differential diagnosis.
In addition, insight into mechanism and etiology complements the differential diagnosis.
 Onset of anemia has a great impact on symptoms and diagnosis.
Onset of anemia has a great impact on symptoms and diagnosis.
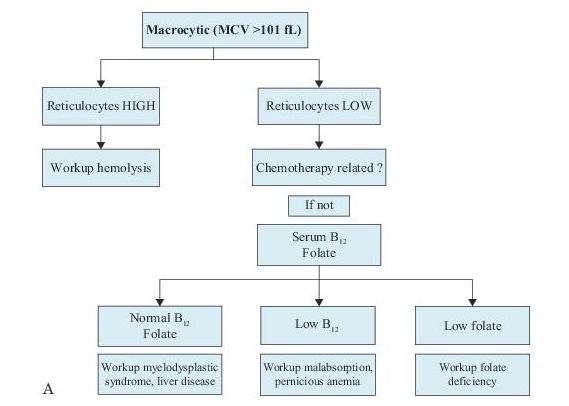
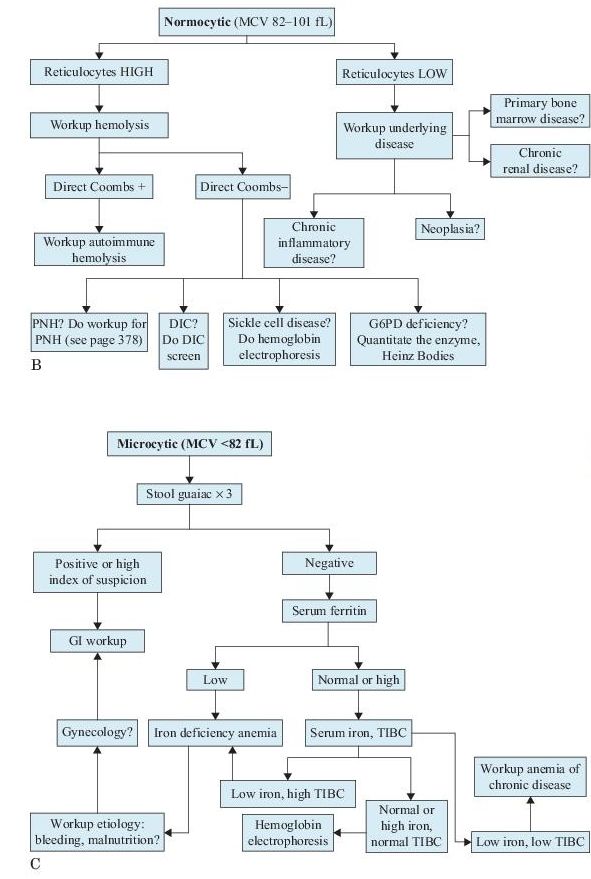
Figure 9–1 Workup of anemias based on the mean corpuscular volume (MCV).
Onset
 Acute
Acute
 Bleeding
Bleeding
 Hemolysis
Hemolysis
 Acute bone marrow disease (e.g., leukemias)
Acute bone marrow disease (e.g., leukemias)
 Chronic
Chronic
 Deficiencies: iron (most common), folic acid, vitamin B12, nutritional
Deficiencies: iron (most common), folic acid, vitamin B12, nutritional
 Congenital (hemoglobinopathies, hereditary spherocytosis)
Congenital (hemoglobinopathies, hereditary spherocytosis)
 Neoplasia, especially metastatic or hematologic malignancies
Neoplasia, especially metastatic or hematologic malignancies
 Renal disease
Renal disease
 Chronic inflammatory disorders
Chronic inflammatory disorders
 Many others
Many others
When to Suspect Anemia
 Children
Children
 Young child who fails to thrive and is not as active as expected for age.
Young child who fails to thrive and is not as active as expected for age.
 Anemia detected at ages 3−6 months suggests a congenital disorder of Hb synthesis or structure.
Anemia detected at ages 3−6 months suggests a congenital disorder of Hb synthesis or structure.
 Adults.
Adults.
 Nonspecific symptoms and signs such as weakness, dizziness, progressive lack of energy, pallor, and shortness of breath in the absence of serious heart or lung disease (overt CHF may develop as a consequence of severe anemia).
Nonspecific symptoms and signs such as weakness, dizziness, progressive lack of energy, pallor, and shortness of breath in the absence of serious heart or lung disease (overt CHF may develop as a consequence of severe anemia).
 Protracted GI or vaginal bleeding.
Protracted GI or vaginal bleeding.
 A family history of anemia.
A family history of anemia.
 Jaundice or red urine.
Jaundice or red urine.
 Laboratory Findings
Laboratory Findings
 Initial laboratory investigation should include a complete CBC with a reticulocyte count and examination of the peripheral blood smear (PBS). The reticulocyte count reflects bone marrow response to anemia.
Initial laboratory investigation should include a complete CBC with a reticulocyte count and examination of the peripheral blood smear (PBS). The reticulocyte count reflects bone marrow response to anemia.
 Once the suspicion of anemia is confirmed by finding a reduction in Hb (the RBC count may be normal or even higher in certain conditions, such a thalassemia trait), the type of anemia must be determined by subsequent laboratory investigations, based mostly on the MCV, and subdivided by pathophysiology.
Once the suspicion of anemia is confirmed by finding a reduction in Hb (the RBC count may be normal or even higher in certain conditions, such a thalassemia trait), the type of anemia must be determined by subsequent laboratory investigations, based mostly on the MCV, and subdivided by pathophysiology.
 The RDW provides a useful measurement of the variation in size of RBCs, indicating the presence of anisocytosis when elevated.
The RDW provides a useful measurement of the variation in size of RBCs, indicating the presence of anisocytosis when elevated.
 Once anemia is documented, subsequent investigations depend on the type of anemia suspected based on indices and the reticulocyte count (see Figure 9-1). More complex laboratory tests or bone marrow biopsy may be indicated to ascertain its precise etiology.
Once anemia is documented, subsequent investigations depend on the type of anemia suspected based on indices and the reticulocyte count (see Figure 9-1). More complex laboratory tests or bone marrow biopsy may be indicated to ascertain its precise etiology.
 Various types of anemias are described subsequently.
Various types of anemias are described subsequently.
 Microcytic
Microcytic
 Macrocytic
Macrocytic
 Normocytic
Normocytic
 Aplastic
Aplastic
 Hemoglobinopathies
Hemoglobinopathies
 Hemolytic anemias
Hemolytic anemias
 Sickle cell
Sickle cell
 HbC, HbD, HbE diseases
HbC, HbD, HbE diseases
 Thalassemias
Thalassemias
Suggested Readings
Beutler E, Waalen J. The definition of anemia: what is the lower limit of normal of the blood hemoglobin concentration? Blood. 2006;107:1747−1750.
Tefferi A. Anemia in adults: a contemporary approach to diagnosis. Mayo Clin Proc. 2003; 78:1274−1280.
MACROCYTIC ANEMIAS
 Definition
Definition
Anemias in which the RBCs are oval macrocytes, with an MCV larger than normal (>101 fL).
 Who Should Be Suspected?
Who Should Be Suspected?
A patient with macrocytic anemia, hypersegmented neutrophils on peripheral blood smear, and symptoms of malabsorption, poor diet, chronic hemolysis without folate supplementation, chemotherapy, or hypothyroidism. Folate deficiency is seen with alcoholism; in third-world countries, it may be associated with sprue-like syndromes. Vitamin B12 (cobalamin) deficiency increases in incidence with aging, and should be searched for, even in the absence of anemia in the elderly with neurologic deficits. Cobalamin and folic acid deficiencies often coexist. Other causes of macrocytic anemias are liver cirrhosis, myelodysplastic syndrome (MDS), azidothymidine (AZT) therapy for AIDS, Down syndrome, and normal newborns.
 Laboratory Findings
Laboratory Findings
Laboratory investigation of macrocytic anemias must differentiate between macrocytic anemias without megaloblastosis and true megaloblastic anemias resulting from vitamin B12 and/or folate deficiency. Megaloblastic anemia is a morphologic definition based on bone marrow examination. B12 deficiency may be the result of pernicious anemia (PA) (lack of intrinsic factor) or may have other etiologies.
 CBC:
CBC:
 Anemia with oval macrocytes, poikilocytosis and anisocytosis, small tear-drop cells
Anemia with oval macrocytes, poikilocytosis and anisocytosis, small tear-drop cells
 High RDW
High RDW
 Thrombocytopenia and leukopenia in severe cases
Thrombocytopenia and leukopenia in severe cases
 Hypersegmented polymorphonuclear cells and giant metamyelocytes in megaloblastic anemias
Hypersegmented polymorphonuclear cells and giant metamyelocytes in megaloblastic anemias
 Reticulocyte count: inadequate for the degree of anemia.
Reticulocyte count: inadequate for the degree of anemia.
 Serum or RBC folate and serum cobalamin are obtained if another etiology is not obvious. The specific metabolites methylmalonic acid and homocysteine accumulate in these deficiencies; they are additional assays and may help discriminate between cobalamin and folate deficiencies and other etiologies for macrocytic anemias. These assays, as well as RBC folate, are more expensive and should be reserved for patients with borderline folate or cobalamin values but strong suspicion of one or the other.
Serum or RBC folate and serum cobalamin are obtained if another etiology is not obvious. The specific metabolites methylmalonic acid and homocysteine accumulate in these deficiencies; they are additional assays and may help discriminate between cobalamin and folate deficiencies and other etiologies for macrocytic anemias. These assays, as well as RBC folate, are more expensive and should be reserved for patients with borderline folate or cobalamin values but strong suspicion of one or the other.
 Serum cobalamin if <200 pg/mL is consistent with vitamin B12 deficiency.
Serum cobalamin if <200 pg/mL is consistent with vitamin B12 deficiency.
 Serum folate if <2 ng/mL is consistent with folate deficiency.
Serum folate if <2 ng/mL is consistent with folate deficiency.
 Serum or urine methylmalonic acid if increased confirms vitamin B12 deficiency. It may be normal in folate deficiency.
Serum or urine methylmalonic acid if increased confirms vitamin B12 deficiency. It may be normal in folate deficiency.
 Homocysteine (total) if elevated is compatible with either cobalamin or folate deficiency. If normal, both can be excluded.
Homocysteine (total) if elevated is compatible with either cobalamin or folate deficiency. If normal, both can be excluded.
 Documentation of cobalamin deficiency does not establish the diagnosis of PA, an autoimmune disease characterized by deficiency of intrinsic factor (IF) and lack of HCl gastric secretion. PA was traditionally diagnosed by the absorption of orally administrated radiolabeled cobalamin, the Schilling test (no longer available in the United States). In its absence, the assays mentioned above are helpful, but not specific for PA. Fifty percent to 70% of PA patients will have positive serum anti-IF antibodies, thus documenting PA (100% specificity). The patients who are negative for IF antibodies cannot be distinguished from non-PA cases of cobalamin malabsorption but will respond to oral vitamin B12 if not PA. Antiparietal antibodies are less sensitive or specific. Recently, chronic Helicobacter pylori infection has been implicated in the etiology of PA and the lack of IF.
Documentation of cobalamin deficiency does not establish the diagnosis of PA, an autoimmune disease characterized by deficiency of intrinsic factor (IF) and lack of HCl gastric secretion. PA was traditionally diagnosed by the absorption of orally administrated radiolabeled cobalamin, the Schilling test (no longer available in the United States). In its absence, the assays mentioned above are helpful, but not specific for PA. Fifty percent to 70% of PA patients will have positive serum anti-IF antibodies, thus documenting PA (100% specificity). The patients who are negative for IF antibodies cannot be distinguished from non-PA cases of cobalamin malabsorption but will respond to oral vitamin B12 if not PA. Antiparietal antibodies are less sensitive or specific. Recently, chronic Helicobacter pylori infection has been implicated in the etiology of PA and the lack of IF.
 Bone marrow aspirate (indicated in very selected cases) may reveal marked red cell hyperplasia and megaloblastic maturation in both vitamin B12 and folate deficiencies. Otherwise, it may uncover other reasons for macrocytosis, such as myelodysplastic syndrome MDS.
Bone marrow aspirate (indicated in very selected cases) may reveal marked red cell hyperplasia and megaloblastic maturation in both vitamin B12 and folate deficiencies. Otherwise, it may uncover other reasons for macrocytosis, such as myelodysplastic syndrome MDS.
 Serum LDH and indirect bilirubin are elevated in folate and vitamin B12 deficiency.
Serum LDH and indirect bilirubin are elevated in folate and vitamin B12 deficiency.
 Limitations
Limitations
 In the presence of coexisting iron deficiency, MCV may not be elevated, even in cases of overt folate or cobalamin deficiency.
In the presence of coexisting iron deficiency, MCV may not be elevated, even in cases of overt folate or cobalamin deficiency.
 Low cobalamin levels develop during pregnancy.
Low cobalamin levels develop during pregnancy.
 One hospital meal may normalize serum folate level (but not RBC).
One hospital meal may normalize serum folate level (but not RBC).
Methylmalonic acid increases in renal insufficiency.
MICROCYTIC ANEMIAS
 Definition
Definition
Anemias characterized by low MCV (<82 fL) and hypochromia. Most common: iron deficiency anemia, to be differentiated from the thalassemias and occasionally from anemia of chronic diseases. Despite the high frequency of iron deficiency anemia, patients should not be treated automatically with iron without determining the cause of the anemia.
 Who Should Be Suspected?
Who Should Be Suspected?
Suspect iron deficiency if the following are present:
 History of GI, vaginal, or massive, repeated urinary bleeding
History of GI, vaginal, or massive, repeated urinary bleeding
 Microcytosis, hypochromic
Microcytosis, hypochromic
 Poor diet
Poor diet
 Laboratory Findings
Laboratory Findings
 First line of investigation: serum ferritin has a specificity of 98% but a sensitivity of only 25% for a 12 μg/L threshold. Because ferritin is an acute-phase reactant, it may be normal or even increased despite iron deficiency when the patients have serious medical problems, such as chronic inflammatory conditions and active liver disease. As a consequence, a normal ferritin value does not exclude iron deficiency. Very low values are definitely diagnostic, iron deficiency is confirmed, and there is no need to obtain serum iron and total iron-binding capacity (TIBC). Investigation of etiology (history, stool examination for occult blood, GI investigation, pelvic and rectal examinations) is mandatory.
First line of investigation: serum ferritin has a specificity of 98% but a sensitivity of only 25% for a 12 μg/L threshold. Because ferritin is an acute-phase reactant, it may be normal or even increased despite iron deficiency when the patients have serious medical problems, such as chronic inflammatory conditions and active liver disease. As a consequence, a normal ferritin value does not exclude iron deficiency. Very low values are definitely diagnostic, iron deficiency is confirmed, and there is no need to obtain serum iron and total iron-binding capacity (TIBC). Investigation of etiology (history, stool examination for occult blood, GI investigation, pelvic and rectal examinations) is mandatory.
 If serum ferritin is normal or borderline, serum iron and transferrin (usually reported as TIBC) are the next assay to be ordered.
If serum ferritin is normal or borderline, serum iron and transferrin (usually reported as TIBC) are the next assay to be ordered.
 If the serum iron is very low and TIBC elevated (with the ratio of serum iron divided by TIBC <16%), diagnosis is confirmed.
If the serum iron is very low and TIBC elevated (with the ratio of serum iron divided by TIBC <16%), diagnosis is confirmed.
 Normal serum iron and TIBC: iron deficiency is excluded in most cases.
Normal serum iron and TIBC: iron deficiency is excluded in most cases.
 Low serum iron, low TIBC: most likely anemia of chronic disease; workup underlying etiology.
Low serum iron, low TIBC: most likely anemia of chronic disease; workup underlying etiology.
 High serum iron, normal TIBC: the most likely diagnosis is thalassemia.
High serum iron, normal TIBC: the most likely diagnosis is thalassemia.
 Two additional blood tests: the soluble transferrin receptor and the reticulocyte Hb content are optional. When used in conjunction with ferritin, these tests improve further our ability to accurately diagnose iron deficiency. Not widely used.
Two additional blood tests: the soluble transferrin receptor and the reticulocyte Hb content are optional. When used in conjunction with ferritin, these tests improve further our ability to accurately diagnose iron deficiency. Not widely used.
 As a last resort, if the diagnosis is still in doubt: bone marrow aspirate/biopsy for Prussian blue stain. If it is negative, iron deficiency is definitely present.
As a last resort, if the diagnosis is still in doubt: bone marrow aspirate/biopsy for Prussian blue stain. If it is negative, iron deficiency is definitely present.
NORMOCYTIC ANEMIAS
 Definition
Definition
Anemias with normal MCV.
 Who Should Be Suspected?
Who Should Be Suspected?
Patients with anemias secondary to an underlying nonhematologic disease (also known as “anemias of chronic disease” [ACD]). The term “anemia of chronic inflammation” may be used too, but it does not cover all situations (see the following paragraph). The most common conditions leading to ACD:
 Anemia of chronic inflammation (infections, rheumatologic diseases) is the prototype of normocytic anemias; the red cell may occasionally be borderline microcytic.
Anemia of chronic inflammation (infections, rheumatologic diseases) is the prototype of normocytic anemias; the red cell may occasionally be borderline microcytic.
 The etiology of anemia of chronic renal failure is in part the reduced production of erythropoietin; additional factors are a shortened red cell survival and frequent bleeding.
The etiology of anemia of chronic renal failure is in part the reduced production of erythropoietin; additional factors are a shortened red cell survival and frequent bleeding.
 Anemia in cancer patients is a common, multifactorial finding. Microangiopathic hemolytic anemia and myelophthisic anemia may be an additional feature resulting from disseminated carcinoma.
Anemia in cancer patients is a common, multifactorial finding. Microangiopathic hemolytic anemia and myelophthisic anemia may be an additional feature resulting from disseminated carcinoma.
 Aplastic anemias (AAs) can be congenital or acquired. In AA, hematopoiesis fails. All blood lineages are decreased (pancytopenia), with the possible exception of lymphocytes. Pure red cell anemia is a variant of AA in which only, or mostly, the red cell line is affected.
Aplastic anemias (AAs) can be congenital or acquired. In AA, hematopoiesis fails. All blood lineages are decreased (pancytopenia), with the possible exception of lymphocytes. Pure red cell anemia is a variant of AA in which only, or mostly, the red cell line is affected.
 Laboratory Findings
Laboratory Findings
 CBC: Moderate anemia, normal to slightly reduced MCV in inflammatory conditions; normal red cell morphology, with only mild variation in RDW. In anemia of chronic renal failure, burr cells can be seen on the peripheral blood smear (PBS).
CBC: Moderate anemia, normal to slightly reduced MCV in inflammatory conditions; normal red cell morphology, with only mild variation in RDW. In anemia of chronic renal failure, burr cells can be seen on the peripheral blood smear (PBS).
 Inadequate reticulocyte response.
Inadequate reticulocyte response.
 Increased serum ferritin; reduced serum iron and TIBC.
Increased serum ferritin; reduced serum iron and TIBC.
 Serum erythropoietin is inadequate for the level of anemia, especially in renal failure.
Serum erythropoietin is inadequate for the level of anemia, especially in renal failure.
APLASTIC ANEMIA (AA)
 Definition
Definition
Although the name refers only to anemia, AA is characterized by peripheral blood pancytopenia. It is the paradigm of bone marrow failure. The diagnosis of AA is one of exclusion. There is variable bone marrow hypocellularity due to diminished or absent hematopoietic precursors, the result of injury to the pluripotent stem cell. The absence of a myeloproliferative neoplasm or an MDS is a prerequisite for the diagnosis.
 Etiology
Etiology
 AA may be acquired or congenital (Fanconi anemia; see below). More than 50% of the acquired cases are idiopathic, most likely due to an autoimmune mechanism that destroys or suppresses the hematopoietic stem cell via cytotoxic T lymphocytes and the cytokines they produce.
AA may be acquired or congenital (Fanconi anemia; see below). More than 50% of the acquired cases are idiopathic, most likely due to an autoimmune mechanism that destroys or suppresses the hematopoietic stem cell via cytotoxic T lymphocytes and the cytokines they produce.
 Other cases may result from drugs, such as chemotherapy, anticonvulsants, and many more. It is essential to obtain a history of drug or toxin exposure.
Other cases may result from drugs, such as chemotherapy, anticonvulsants, and many more. It is essential to obtain a history of drug or toxin exposure.
 Immunologic disorders such as graft versus host disease.
Immunologic disorders such as graft versus host disease.
 Thymomas.
Thymomas.
 Exposure to ionizing radiation.
Exposure to ionizing radiation.
 Viral infections: EBV and the putative agent of seronegative hepatitis.
Viral infections: EBV and the putative agent of seronegative hepatitis.
 Severe malnutrition: kwashiorkor, anorexia nervosa.
Severe malnutrition: kwashiorkor, anorexia nervosa.
 Leukemia may be the underlying disease in 1−5% of patients who present with AA.
Leukemia may be the underlying disease in 1−5% of patients who present with AA.
 PNH (see p. 378) develops in 5−10% of patients with AA; conversely, AA develops in 25% of patients with PNH.
PNH (see p. 378) develops in 5−10% of patients with AA; conversely, AA develops in 25% of patients with PNH.
 Who Should Be Suspected?
Who Should Be Suspected?
An individual who presents with a clinical picture of increasing symptoms of anemia, mucosal bleeding, or fever, mucosal ulcerations, and bacterial infections due to neutropenia, in whom an initial CBC demonstrates pancytopenia. Pancytopenia from other causes, such as chemotherapy, should be ruled out (see below). The disease is frequent in East Asia.
 Laboratory Findings
Laboratory Findings
 RBC: anemia is normocytic, normochromic. Hg may be <7 g/L. RDW is normal. MCV is frequently elevated at presentation.
RBC: anemia is normocytic, normochromic. Hg may be <7 g/L. RDW is normal. MCV is frequently elevated at presentation.
 Reticulocytes are invariably decreased to absent.
Reticulocytes are invariably decreased to absent.
 WBC: neutropenia (absolute neutrophil count <1,500/μL) is always present, often accompanied by monocytosis. Abnormal WBCs are not seen. Lymphocyte count is normal (false lymphocytosis if one observes the percent of WBC rather than the absolute count).
WBC: neutropenia (absolute neutrophil count <1,500/μL) is always present, often accompanied by monocytosis. Abnormal WBCs are not seen. Lymphocyte count is normal (false lymphocytosis if one observes the percent of WBC rather than the absolute count).
 Platelets are decreased, but severity varies.
Platelets are decreased, but severity varies.
 Bone marrow (BM) is hypocellular, with an “empty” marrow in severe cases. Less than 30% of residual cells are hematopoietic. Hematopoiesis is not megaloblastic. The appearance of the BM in inherited or acquired AA is identical. Aspiration and biopsy are both necessary to rule out MDS, leukemias, granulomatous disease, or tumors. The BM examination must also exclude the viral hemophagocytic syndrome.
Bone marrow (BM) is hypocellular, with an “empty” marrow in severe cases. Less than 30% of residual cells are hematopoietic. Hematopoiesis is not megaloblastic. The appearance of the BM in inherited or acquired AA is identical. Aspiration and biopsy are both necessary to rule out MDS, leukemias, granulomatous disease, or tumors. The BM examination must also exclude the viral hemophagocytic syndrome.
 Cytogenetics: normal karyotype.
Cytogenetics: normal karyotype.
 Flow cytometry phenotyping shows virtual absence of CD34 hematopoietic stem cells in blood and marrow. AA and PNH overlap in approximately 40−50% of cases.
Flow cytometry phenotyping shows virtual absence of CD34 hematopoietic stem cells in blood and marrow. AA and PNH overlap in approximately 40−50% of cases.
 Serum iron studies are normal.
Serum iron studies are normal.
Suggested Reading
Sheinberg P, Young N. How I treat acquired aplastic anemia. Blood. 2012;120:1185−1196.
PANCYTOPENIA
 Definition
Definition
Pancytopenia is a disorder in which all three blood lines, red cells, white cells, and platelets, are reduced. It is not a disease entity, but a triad of findings that may result from a number of disease processes, most of which involve the bone marrow. Occasionally early on, only two of the three blood lines may be decreased: bycytopenia. Eventually, all three lineages become affected.
 Etiology
Etiology
Pancytopenia may be the result of a congenital anomaly, neoplasia, or autoimmunity or may be iatrogenic (Figure 9-1). The following mechanisms may account for pancytopenia:
 Decreased production of hematopoietic cells by the bone marrow resulting in a hypocellular marrow;
Decreased production of hematopoietic cells by the bone marrow resulting in a hypocellular marrow;
 Ineffective hematopoiesis with a cellular (or even hypercellular) marrow;
Ineffective hematopoiesis with a cellular (or even hypercellular) marrow;
 Infiltration of the bone marrow by extraneous elements;
Infiltration of the bone marrow by extraneous elements;
 Systemic conditions.
Systemic conditions.
A bone marrow aspirate and biopsy are mandatory in most cases without a clear etiology. Deciding the bone marrow cellularity may at times be difficult, because of imprecise quantitation of cellularity or sample error due to unequal distribution of bone marrow tissue. In some cases, biopsies from multiple sites may be necessary. Moreover, hypocellular marrow due to aplastic anemia may evolve over time into a hypercellular marrow. This happens for instance when acute leukemia or PNH develops.
A thorough history and physical examination also play a prominent role in establishing the etiology of pancytopenia, with important clues, such as a history of any drug or toxin exposure or splenomegaly, directing the clinician to possible etiologic causes.
When to suspect pancytopenia:
 Finding a persistent decrease in all three hematopoietic lines on a routine CBC
Finding a persistent decrease in all three hematopoietic lines on a routine CBC
 Clinical symptoms suggestive of anemia, bleeding, or prolonged fever
Clinical symptoms suggestive of anemia, bleeding, or prolonged fever
 Repeated infections
Repeated infections
Tests recommended:
 CBC with differential.
CBC with differential.
 Chemistry, immunology, or infectious investigations as suggested by systemic manifestations.
Chemistry, immunology, or infectious investigations as suggested by systemic manifestations.
 Flow cytometry to rule out paroxysmal nocturnal hemoglobinuria (PNH) or hematologic malignancies.
Flow cytometry to rule out paroxysmal nocturnal hemoglobinuria (PNH) or hematologic malignancies.
 Bone marrow aspiration and biopsy (see above).
Bone marrow aspiration and biopsy (see above).
 Cytogenetic and FISH analysis may establish the precise diagnosis in myelodysplastic syndromes or other hematologic malignancies. Newer whole-genome scanning technologies such as single nucleotide polymorphism (SNP) array–based karyotyping may be an additional diagnostic technology.
Cytogenetic and FISH analysis may establish the precise diagnosis in myelodysplastic syndromes or other hematologic malignancies. Newer whole-genome scanning technologies such as single nucleotide polymorphism (SNP) array–based karyotyping may be an additional diagnostic technology.
 Histochemistry for infiltrative congenital disorders.
Histochemistry for infiltrative congenital disorders.
Suggested Readings
Nester CM, Thomas CP. Atypical hemolytic uyremic syndrome: what it is, how is it diagnosed, and how is it treated. Hematology Am Soc Hematol Educ Program. 2012;2012: 617–625.
Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012;120:1185–1196.
PURE RED CELL APLASIA (PRCA)
 Definition
Definition
Chronic condition of profound anemia, characterized by severe reduction or absence of reticulocytes, and absent bone marrow erythroid precursors. (The congenital pure red cell aplasia is described below under Diamond-Blackfan anemia.) All other cell lines are normal. Most cases are mediated by IgG autoantibodies. PRCA may be associated with certain drugs, thymomas, collagen vascular syndromes, or CLL, or follow parvovirus B19 infection. It may also be part of the 5q−myelodysplastic syndrome. PRCA may also develop following administration of recombinant erythropoietin due to the development of antierythropoietin antibodies.
 Laboratory Findings
Laboratory Findings
 CBC: severe reduction but normal-appearing RBCs; normal WBC and platelet counts.
CBC: severe reduction but normal-appearing RBCs; normal WBC and platelet counts.
 Reticulocytes are severely decreased or absent.
Reticulocytes are severely decreased or absent.
 Bone marrow is normocellular, but erythroid precursors cells are absent (giant normoblasts may be seen if the etiology is a parvovirus infection). White cell precursors and megakaryocytes (except in the 5q− syndrome) are normal.
Bone marrow is normocellular, but erythroid precursors cells are absent (giant normoblasts may be seen if the etiology is a parvovirus infection). White cell precursors and megakaryocytes (except in the 5q− syndrome) are normal.
 Serum iron and transferrin saturation are increased.
Serum iron and transferrin saturation are increased.
FANCONI ANEMIA (FA)
 Definition
Definition
The most common inherited AA. Autosomal recessive syndrome in childhood, associated with congenital anomalies of short stature, rudimentary thumbs, hypoplastic radii, renal anomalies, and skin spots. There is an increased incidence of myelodysplastic syndrome, acute myelogenous leukemia, and squamous cell carcinomas. The diagnosis is usually made between the ages of 6 and 9 years, but in rare cases, it may not been made until adulthood.
 Laboratory Findings
Laboratory Findings
The hematologic findings evolve over months or years: macrocytic anemia, leukopenia due to neutropenia, and mild to moderate thrombocytopenia.
 Cytogenetics: Normal chromosome numbers but structural instability causing breaks, gaps, constrictions, and rearrangements. The diagnosis is made by the presence of increased chromosomal breakage in lymphocytes cultured in the presence of DNA cross-linking agents.
Cytogenetics: Normal chromosome numbers but structural instability causing breaks, gaps, constrictions, and rearrangements. The diagnosis is made by the presence of increased chromosomal breakage in lymphocytes cultured in the presence of DNA cross-linking agents.
 Genetics: Multiple genes appear to be responsible for Fanconi anemia. The genes are dispersed through the genome.
Genetics: Multiple genes appear to be responsible for Fanconi anemia. The genes are dispersed through the genome.
 Fetal Hb is increased (>28%).
Fetal Hb is increased (>28%).
 i antigen may be observed.
i antigen may be observed.
 Serum alpha-protein levels are frequently elevated.
Serum alpha-protein levels are frequently elevated.
DIAMOND-BLACKFAN ANEMIA (DBA)
 Definition
Definition
DBA is a congenital pure red cell aplasia. It is usually sporadic but may be inherited in an autosomal dominant manner. Onset is before 12 months. DBA is associated with congenital anomalies of the kidneys, eyes, skeleton, and heart. Spontaneous remissions have been observed in 20–30% of cases after months or years.
 Laboratory Findings
Laboratory Findings
 RBC: severe macrocytic, anemia that is refractory to conventional therapies.
RBC: severe macrocytic, anemia that is refractory to conventional therapies.
 Reticulocytes are <1%.
Reticulocytes are <1%.
 WBC, differential white cell count, and platelet count are normal.
WBC, differential white cell count, and platelet count are normal.
 Bone marrow is normocellular but presents with a marked decrease in erythroid precursors. All other cell lines are normal.
Bone marrow is normocellular but presents with a marked decrease in erythroid precursors. All other cell lines are normal.
 Fetal Hb is increased.
Fetal Hb is increased.
 Adenosine deaminase is increased in RBCs.
Adenosine deaminase is increased in RBCs.
 Serum iron and all other hematologic parameters are normal.
Serum iron and all other hematologic parameters are normal.
 Serum erythropoietin is elevated.
Serum erythropoietin is elevated.
 HEMOGLOBINOPATHIES
HEMOGLOBINOPATHIES
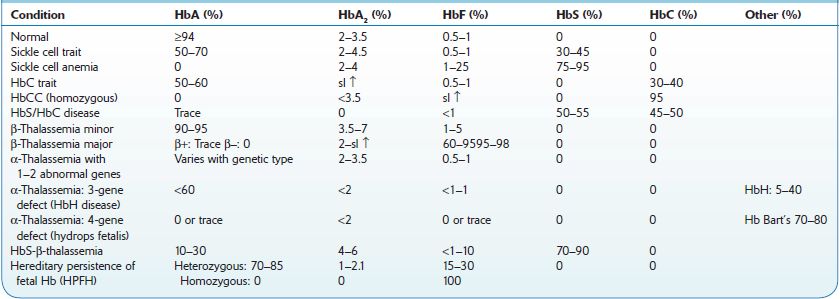
Hemoglobinopathies constitute the most common inherited disorders in humans as a result of selective pressure of endemic falciparum malaria. Human hemoglobins (Hb) are proteins containing a heme moiety and two pairs of globin genes. Normal adult Hb is composed of two alpha and two β-chains, which together add up to 97% of total Hb in red cells (RBC). The balance globins are composed of Hb A2 (approximately 2.5%) and fetal Hg (HbF) usually 0.8–2%. More than 1,000 mutations involving the globin genes have been described; they result from amino acid substitution or from abnormalities of synthesis. The majority of these variants do not cause clinical or hematologic problems. Several variants, such as sickle cell disease and β-thalassemias (described below), are protective and asymptomatic in the heterozygous; however, they result in severe morbidity in the homozygous. Initial screening and definitive diagnosis for Hb variants are described in Chapter 16. Table 9-1 describes the most common hemoglobinopathies encountered in North America: sickle cell syndromes, HbC disease, and β- and α-thalassemias. Genetic analysis may be necessary for uncommon or unknown variants. In North America, it is done in a few specialized laboratories.
TABLE 9–1. Hemoglobinopathies
SICKLE CELL ANEMIA
 Definition
Definition
The term sickle cell disease (SCD) describes all the conditions associated with sickling of RBC. SCD encompasses a group of conditions with autosomal inheritance of abnormal Hb β chain resulting from a substitution of valine for glutamic acid in the beta globin chain. This substitution results in polymerization of poorly soluble deoxy-HbS, leading to a marked decrease in red cells’ deformability and irreversible distortion of red cells into the sickle cell shape, with their removal by the spleen (prior to the occurrence of autosplenectomy) and macrophages. SCD is encountered mostly in populations of African or Arab ancestry, as well as in some Indian groups.
 Sickle cell anemia (SCA) is the homozygous state where the majority of Hb is S. This results in the precipitation and polymerization of Hb, causing rigid crystals that deform red cells (sickling), leading to microvascular occlusions and hemolysis.
Sickle cell anemia (SCA) is the homozygous state where the majority of Hb is S. This results in the precipitation and polymerization of Hb, causing rigid crystals that deform red cells (sickling), leading to microvascular occlusions and hemolysis.
 Sickle cell trait (SCT) is the heterozygous form, in which the CBC is normal. Although generally asymptomatic, its diagnosis is important for genetic counseling.
Sickle cell trait (SCT) is the heterozygous form, in which the CBC is normal. Although generally asymptomatic, its diagnosis is important for genetic counseling.
 Sickle cell syndromes (diseases) represent combinations of sickle cell trait with other hemoglobinopathies, most commonly with β-thalassemia or Hb C.
Sickle cell syndromes (diseases) represent combinations of sickle cell trait with other hemoglobinopathies, most commonly with β-thalassemia or Hb C.
 Who Should Be Suspected?
Who Should Be Suspected?
 SCA should be suspected in a child with a family history of sickle cell disease, failure to thrive, progressive hemolytic anemia, and vasoocclusive crises (repeated painful episodes that lead to organ damage).
SCA should be suspected in a child with a family history of sickle cell disease, failure to thrive, progressive hemolytic anemia, and vasoocclusive crises (repeated painful episodes that lead to organ damage).
 Clinical manifestations are not present at birth, but become apparent after 3–6 months of life, as the concentration of HbF declines, and that of HbS increases. By 2 years of age, 61% of children have already had painful vasoocclusive episodes.
Clinical manifestations are not present at birth, but become apparent after 3–6 months of life, as the concentration of HbF declines, and that of HbS increases. By 2 years of age, 61% of children have already had painful vasoocclusive episodes.
 Aplastic crises are self-limited episodes of erythroid aplasia lasting 5–10 days. They are due to infections (most commonly parvovirus B19) and may require emergency transfusions.
Aplastic crises are self-limited episodes of erythroid aplasia lasting 5–10 days. They are due to infections (most commonly parvovirus B19) and may require emergency transfusions.
 Bilirubin gallstones are present in 30% of patients by age 18 and 70% by age 30.
Bilirubin gallstones are present in 30% of patients by age 18 and 70% by age 30.
 Organ damage develops by the time SCA patients are in their teens, with the lungs, kidneys, heart, and liver involvement. Cerebrovascular accidents are also common.
Organ damage develops by the time SCA patients are in their teens, with the lungs, kidneys, heart, and liver involvement. Cerebrovascular accidents are also common.
 Laboratory Findings
Laboratory Findings
 A “sickle cell screen” can be obtained for a rapid preliminary diagnosis. It is positive in SCA, SCT, in some non-S sickling hemoglobinopathies, and in combined SCD with other hemoglobinopathies.
A “sickle cell screen” can be obtained for a rapid preliminary diagnosis. It is positive in SCA, SCT, in some non-S sickling hemoglobinopathies, and in combined SCD with other hemoglobinopathies.
 Hb variant analysis (HPLC or electrophoresis) is used to identify different hemoglobins. Newborns have predominantly HbF with a small amount of HbS and no HbA1. Because other sickle cell syndromes may have similar patterns, it is recommended to study the parents, or repeat the test after 1 year of age, when the adult pattern of SCA is established: very high HbS. HbF may be slightly elevated (1–4%) and especially in patients treated successfully with hydroxyurea where it may reach 15% or more, resulting in marked diminution in morbidity.
Hb variant analysis (HPLC or electrophoresis) is used to identify different hemoglobins. Newborns have predominantly HbF with a small amount of HbS and no HbA1. Because other sickle cell syndromes may have similar patterns, it is recommended to study the parents, or repeat the test after 1 year of age, when the adult pattern of SCA is established: very high HbS. HbF may be slightly elevated (1–4%) and especially in patients treated successfully with hydroxyurea where it may reach 15% or more, resulting in marked diminution in morbidity.
 The newborn with SCT will have HbA, HbF, and HbS. Adults have >50% HbA1 and 35–45% HbS.
The newborn with SCT will have HbA, HbF, and HbS. Adults have >50% HbA1 and 35–45% HbS.
 Prenatal testing: gene analysis of fetal DNA may be performed on chorionic villi (7–10 weeks of gestation) or amniocytes (15–20 weeks of gestation). DNA testing may be also useful in newborns or children in cases with high levels of HbF if hereditary persistence of fetal hemoglobin is suspected.
Prenatal testing: gene analysis of fetal DNA may be performed on chorionic villi (7–10 weeks of gestation) or amniocytes (15–20 weeks of gestation). DNA testing may be also useful in newborns or children in cases with high levels of HbF if hereditary persistence of fetal hemoglobin is suspected.
 Patients with HbSC disease (see below) have equal amount of HbS and C.
Patients with HbSC disease (see below) have equal amount of HbS and C.
 Patients with sickle cell trait–β-thalassemia (+) have HbA1, elevated HbA2, and HbS.
Patients with sickle cell trait–β-thalassemia (+) have HbA1, elevated HbA2, and HbS.
 CBC in patients with SCA.
CBC in patients with SCA.
 RBC: mild to moderate chronic hemolytic anemia (Hct 15–30%, Hb 5–10 g/dL), punctuated by aplastic crises (sudden, life-threatening episodes of very severe anemia) (see above).
RBC: mild to moderate chronic hemolytic anemia (Hct 15–30%, Hb 5–10 g/dL), punctuated by aplastic crises (sudden, life-threatening episodes of very severe anemia) (see above).
 Reticulocytes 3–15% (they may account for an elevated MCV).
Reticulocytes 3–15% (they may account for an elevated MCV).
 MCV is in general normal (except as noted above); MCHC is elevated. Microcytosis and hypochromia may be present, however, if there is coexisting α- or β-thalassemia, or iron deficiency in nontransfused patients.
MCV is in general normal (except as noted above); MCHC is elevated. Microcytosis and hypochromia may be present, however, if there is coexisting α- or β-thalassemia, or iron deficiency in nontransfused patients.
 Peripheral blood smear (PBS): visible sickle cells, polychromasia, and Howell-Jolly bodies in older children, reflecting hyposplenism due to autosplenectomy. Nucleated red cells, basophilic stippling, and Pappenheimer bodies are usually found.
Peripheral blood smear (PBS): visible sickle cells, polychromasia, and Howell-Jolly bodies in older children, reflecting hyposplenism due to autosplenectomy. Nucleated red cells, basophilic stippling, and Pappenheimer bodies are usually found.
 WBCs may be higher than normal. A persistent leukocytosis augurs a poor prognosis.
WBCs may be higher than normal. A persistent leukocytosis augurs a poor prognosis.
 Platelets may be elevated, in part the result of loss of splenic function.
Platelets may be elevated, in part the result of loss of splenic function.
 Bone marrow aspirate (not necessary for diagnosis) is hyperplastic.
Bone marrow aspirate (not necessary for diagnosis) is hyperplastic.
 Serum erythropoietin may be inappropriately low in some patients, possibly as the result of progressive renal disease.
Serum erythropoietin may be inappropriately low in some patients, possibly as the result of progressive renal disease.
 Serum iron and ferritin may be low, and transferrin elevated, due to iron loss in urine.
Serum iron and ferritin may be low, and transferrin elevated, due to iron loss in urine.
 Serum folate is low due to overutilization, if not replaced therapeutically.
Serum folate is low due to overutilization, if not replaced therapeutically.
 Serum LDH is elevated.
Serum LDH is elevated.
 Serum bilirubin is commonly elevated.
Serum bilirubin is commonly elevated.
 Serum haptoglobin is decreased.
Serum haptoglobin is decreased.
 Serum aminotransferase is often elevated.
Serum aminotransferase is often elevated.
 Ferritin becomes very elevated in multiply transfused patients.
Ferritin becomes very elevated in multiply transfused patients.
 Urine hemosiderin and urobilinogen are present (not necessary for diagnosis).
Urine hemosiderin and urobilinogen are present (not necessary for diagnosis).
HEMOGLOBIN S–HEMOGLOBIN C DISEASE
 Definition
Definition
A moderately severe sickling disease, clinically intermediate between sickle cell anemia and sickle cell trait. Occurs in 1 of 833 people of African ancestry.
 Laboratory Findings
Laboratory Findings
 Hb electrophoresis: HbA is absent; HbS and HbC are present in approximately equal amounts. HbF is ≤6%.
Hb electrophoresis: HbA is absent; HbS and HbC are present in approximately equal amounts. HbF is ≤6%.
 CBC.
CBC.
 Anemia: mild to moderate normochromic, normocytic.
Anemia: mild to moderate normochromic, normocytic.
 Peripheral blood smear (PBS): tetragonal crystals within the RBC in 70% of patients. Target cells and plump/angulated sickle cells, rather than typical sickle cells, are identified.
Peripheral blood smear (PBS): tetragonal crystals within the RBC in 70% of patients. Target cells and plump/angulated sickle cells, rather than typical sickle cells, are identified.
 MCV is low, or low normal; MCHC is high.
MCV is low, or low normal; MCHC is high.
SICKLE CELL–α-THALASSEMIA DISEASE
α-Thalassemia modifies the severity of sickle cell anemia. Otherwise, it is usually clinically insignificant.
SICKLE CELL–β-THALASSEMIA DISEASE
 Definition
Definition
Condition of mild to moderate severity found in 1 of every 1,667 people of African ancestry.
 Laboratory Findings
Laboratory Findings
 Hb electrophoresis: HbS varies between 20% and 90%; HbF between 2% and 20%. If the HbS is very high and HbA1 is suppressed, the disease is severe. In milder cases, HbA1 is 25–50%. HbA2 is increased (due to the presence of β-thalassemia), but it has to be differentiated from HbC, which has a similar migration pattern.
Hb electrophoresis: HbS varies between 20% and 90%; HbF between 2% and 20%. If the HbS is very high and HbA1 is suppressed, the disease is severe. In milder cases, HbA1 is 25–50%. HbA2 is increased (due to the presence of β-thalassemia), but it has to be differentiated from HbC, which has a similar migration pattern.
 CBC
CBC
 RBC: hypochromic, microcytic anemia with decreased MCV (iron deficiency must be ruled out).
RBC: hypochromic, microcytic anemia with decreased MCV (iron deficiency must be ruled out).
 Peripheral blood smear (PBS): target cells are prominent; other findings resemble those of sickle cell anemia.
Peripheral blood smear (PBS): target cells are prominent; other findings resemble those of sickle cell anemia.
SICKLE CELL–PERSISTENT HIGH FETAL HEMOGLOBIN
 Definition
Definition
Condition seen in 1 in 25,000 African Americans but also frequent in Arab populations. May be mimicked in patients with sickle cell anemia responding to hydroxyurea therapy. Clinical picture and findings intermediate between sickle cell anemia and trait.
 Laboratory Findings
Laboratory Findings
 Hb electrophoresis: HbF is 20–40%; HbA1 and A2 are absent; HgS is approximately 65%.
Hb electrophoresis: HbF is 20–40%; HbA1 and A2 are absent; HgS is approximately 65%.
 RBC: HbF is unevenly distributed among RBC.
RBC: HbF is unevenly distributed among RBC.
SICKLE CELL–HEMOGLOBIN D DISEASE
 Definition
Definition
Condition resembling HbS/HbC disease; less severe than sickle cell anemia. Found in 1 in 20,000 individuals of African ancestry. Clinically a mild syndrome.
 Laboratory Findings
Laboratory Findings
 Intermediate between those of sickle cell anemia and sickle cell trait.
Intermediate between those of sickle cell anemia and sickle cell trait.
 Hb electrophoresis cannot distinguish HbS from HbD at alkaline pH but can be separated at pH 6.2.
Hb electrophoresis cannot distinguish HbS from HbD at alkaline pH but can be separated at pH 6.2.
Suggested Readings
Vichinsky EP, Mahoney DH Jr. Diagnosis of sickle cell syndromes. UpToDate. In: Basow DS (ed). Waltham, MA: UpToDate, Inc.; 2013.
Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115:5300–5311.
HEMOGLOBIN C DISEASE
 Definition
Definition
A hemoglobinopathy prevalent in individuals with ancestral roots in West Africa.
Autosomal transmission.
 HbC trait: found in 2% of African Americans, less frequently in other groups; asymptomatic, no anemia.
HbC trait: found in 2% of African Americans, less frequently in other groups; asymptomatic, no anemia.
 Homozygous HbC disease: mild hemolytic anemia.
Homozygous HbC disease: mild hemolytic anemia.
 Laboratory Findings
Laboratory Findings
 HbC trait: Hb variant analysis shows 50% HbA1 and 30–40% HbC.
HbC trait: Hb variant analysis shows 50% HbA1 and 30–40% HbC.
 Homozygous condition: There is no HbA1, and HbC forms the majority variant Hb; HgF is slightly increased. Peripheral blood smear (PBS) shows a variable number of target cells (≤40%), a variable number of microspherocytes, occasionally nucleated RBCs, and a few tetragonal crystals within RBCs.
Homozygous condition: There is no HbA1, and HbC forms the majority variant Hb; HgF is slightly increased. Peripheral blood smear (PBS) shows a variable number of target cells (≤40%), a variable number of microspherocytes, occasionally nucleated RBCs, and a few tetragonal crystals within RBCs.
HEMOGLOBIN C–β-THALASSEMIA
HbC-β-thalassemia is a form of β-thalassemia (see below). Affected individuals are commonly asymptomatic, although moderate hemolysis may be present. These individuals have a moderate microcytic, hypochromic, hemolytic anemia, and splenomegaly. Their red cells may show Hb C crystals.
HEMOGLOBIN D DISEASE
 Definition
Definition
Autosomal inherited hemoglobinopathy prevalent in Southeast Asia and in parts of India (HbD Punjab). The heterozygous form is asymptomatic with no anemia.
 Laboratory Findings
Laboratory Findings
 Hb variant analysis demonstrates the abnormal Hb at acid pH (it has the same mobility as HbS at alkaline pH). There are no other laboratory abnormalities in the heterozygous individual.
Hb variant analysis demonstrates the abnormal Hb at acid pH (it has the same mobility as HbS at alkaline pH). There are no other laboratory abnormalities in the heterozygous individual.
 RBC: mild hemolytic, microcytic anemia in homozygous individuals; their peripheral blood smear (PBS) shows target cells and spherocytes.
RBC: mild hemolytic, microcytic anemia in homozygous individuals; their peripheral blood smear (PBS) shows target cells and spherocytes.
HEMOGLOBIN E DISEASE
 Definition
Definition
The most common structural hemoglobinopathy in the United States after HbS and HbC. Autosomal inherited hemoglobinopathy, prevalent in Southeast Asia (15–30% of the population in Cambodia, Thailand, parts of China, Burma, and Vietnam). Heterozygous individuals have similar findings as patients with β-thalassemia trait (see below). Homozygotes exhibit more microcytosis but are asymptomatic.
 Laboratory Findings
Laboratory Findings
 Hb variant analysis shows 95−97% HbE in the homozygous (the rest is HbF); 30−35% in individuals carrying HbE trait. Electrophoretic mobility is the same as for HbA2, but it is present in much higher concentrations. It separates from HbC and O on citrate agar electrophoresis at acid pH.
Hb variant analysis shows 95−97% HbE in the homozygous (the rest is HbF); 30−35% in individuals carrying HbE trait. Electrophoretic mobility is the same as for HbA2, but it is present in much higher concentrations. It separates from HbC and O on citrate agar electrophoresis at acid pH.
 CBC.
CBC.
 Mild hemolytic, microcytic (MCV 55−70 fL) anemia or no anemia in the homozygous.
Mild hemolytic, microcytic (MCV 55−70 fL) anemia or no anemia in the homozygous.
 Erythrocytosis may be present (RBC approximately 5,500/μL) in both the trait and in the homozygous.
Erythrocytosis may be present (RBC approximately 5,500/μL) in both the trait and in the homozygous.
 Peripheral blood smear (PBS) shows 25–60% target cells and microcytes in the homozygous individuals.
Peripheral blood smear (PBS) shows 25–60% target cells and microcytes in the homozygous individuals.
HEMOGLOBIN E–β-THALASSEMIA
 Definition
Definition
The most common symptomatic thalassemia in Southeast Asia. A severe condition that resembles β-thalassemia intermedia or β-thalassemia major (see below).
 Laboratory Findings
Laboratory Findings
 Hemolytic anemia varies from moderate to severe, similar to β-thalassemias (see below).
Hemolytic anemia varies from moderate to severe, similar to β-thalassemias (see below).
 Peripheral blood smear (PBS) shows severe hypochromia and macrocytosis and marked anisopoikilocytosis with many teardrop and target red cells. Nucleated RBC and basophilic stippling may be present.
Peripheral blood smear (PBS) shows severe hypochromia and macrocytosis and marked anisopoikilocytosis with many teardrop and target red cells. Nucleated RBC and basophilic stippling may be present.
HEMOGLOBIN E–ALPHA-THALASSEMIA
A mild hemolytic anemia encountered in Southeast Asia. It causes microcytosis. The severity depends on the number of α genes deleted (see α-thalassemia below).
 THE THALASSEMIAS
THE THALASSEMIAS
Thalassemias are chronic, microcytic, hemolytic anemias. They result from defective synthesis of either β- or α-globin subunits of the Hb A molecule. Thalassemias are classified into β- or α-thalassemia according to which of the globin chain is affected. The thalassemias are among the most common genetic disorders world-wide. They have an autosomal recessive inheritance resulting in either homozygous (thalassemia major) or subtle (thalassemia minor) clinical abnormalities. The β-thalassemia syndromes are extremely heterogenous. In addition to β-thalassemia trait and β-thalassemia major described below, there are combinations with other hemoglobinopathies and variants described above.
β-THALASSEMIA MAJOR
 Definition and Who Should Be Suspected
Definition and Who Should Be Suspected
A severe condition resulting from impaired or absent production of the β globin chains of Hb. The resulting excess α chains precipitate inside the red cells with dire consequences: severe hemolysis, skeletal changes, liver abnormalities, premature gallbladder bilirubin stones, splenomegaly, aplastic crises, impaired growth, endocrine and cardiopulmonary complications, and hemosiderosis resulting from RBC transfusions. The clinical expression of the severe phenotype is extremely heterogenous. A milder form of β-thalassemia, b-thalassemia intermedia, is seen in patients with one β (−) allele mutation that produce no β globin chains and with a β (+) mutation from the second allele. It produces a small amount of β chains, thus these patients are less severely affected.
 β-thalassemia is most common in individuals of Mediterranean ancestry (mutations result from protection against endemic malaria in the Mediterranean basin); it is also found in African Americans and in some groups in India.
β-thalassemia is most common in individuals of Mediterranean ancestry (mutations result from protection against endemic malaria in the Mediterranean basin); it is also found in African Americans and in some groups in India.
 Infants are well at birth, depending on high levels of HbF (no β chains, just α and fetal globins), for tissue oxygenation. The diagnosis is usually established at 6–12 months of age due to increasing symptoms: pallor, irritability, growth retardation abdominal swelling due to hepatosplenomegaly, followed by abnormal skeletal development, the result of an expanding extramedullary hematopoiesis.
Infants are well at birth, depending on high levels of HbF (no β chains, just α and fetal globins), for tissue oxygenation. The diagnosis is usually established at 6–12 months of age due to increasing symptoms: pallor, irritability, growth retardation abdominal swelling due to hepatosplenomegaly, followed by abnormal skeletal development, the result of an expanding extramedullary hematopoiesis.
 Coinheritance of an α-thalassemia trait may ameliorate the morbidity of β-thalassemia major.
Coinheritance of an α-thalassemia trait may ameliorate the morbidity of β-thalassemia major.
 Laboratory Findings
Laboratory Findings
 CBC.
CBC.
 RBC: profound anemia, microcytosis, reduced MCV and MCHC, very elevated RDW. Hb levels may be as low as 3–4 g/dL. The anemia may become acutely life threatening during aplastic crises, mostly provoked by infection with parvovirus B19, which infects precursor erythroid cells.
RBC: profound anemia, microcytosis, reduced MCV and MCHC, very elevated RDW. Hb levels may be as low as 3–4 g/dL. The anemia may become acutely life threatening during aplastic crises, mostly provoked by infection with parvovirus B19, which infects precursor erythroid cells.
 Red cell morphology shows extreme hypochromia and poikilocytosis, tear drop cells, and many target cells. Heinz bodies are readily identified when the smears are stained with supravital stains.
Red cell morphology shows extreme hypochromia and poikilocytosis, tear drop cells, and many target cells. Heinz bodies are readily identified when the smears are stained with supravital stains.
 WBC is elevated (in part falsely so, due to enumeration of nucleated RBCs as WBCs by some automated counters), but true leukocytosis is usually present.
WBC is elevated (in part falsely so, due to enumeration of nucleated RBCs as WBCs by some automated counters), but true leukocytosis is usually present.
 Platelets may be reduced due to hypersplenism but become elevated in splenectomized patients.
Platelets may be reduced due to hypersplenism but become elevated in splenectomized patients.
 Peripheral blood smear (PBS): marked poikilocytosis with many target cells, tear drop cells, nucleated RBCs, and basophilic stippling of RBCs.
Peripheral blood smear (PBS): marked poikilocytosis with many target cells, tear drop cells, nucleated RBCs, and basophilic stippling of RBCs.
 Reticulocyte count is inappropriately low, in part the result of ineffective erythropoiesis. It may become 0 during aplastic crises.
Reticulocyte count is inappropriately low, in part the result of ineffective erythropoiesis. It may become 0 during aplastic crises.
 Bone marrow aspirate shows red cell hyperplasia with marked shift to early red cell progenitors due to intramedullary hemolysis, in turn the result of accelerated apoptosis. Megaloblastic morphology may be observed in the absence of folate supplements. Extramedullary hematopoiesis develops in the skeletal bones, liver, and spleen.
Bone marrow aspirate shows red cell hyperplasia with marked shift to early red cell progenitors due to intramedullary hemolysis, in turn the result of accelerated apoptosis. Megaloblastic morphology may be observed in the absence of folate supplements. Extramedullary hematopoiesis develops in the skeletal bones, liver, and spleen.
 Hb variant analysis shows absence of HbA1 in β(0) thalassemia, where only HbA2 and HbF are present. HbA2 may increase to 3–6% (unless iron deficiency is also present). HbA1 is present after RBC transfusions.
Hb variant analysis shows absence of HbA1 in β(0) thalassemia, where only HbA2 and HbF are present. HbA2 may increase to 3–6% (unless iron deficiency is also present). HbA1 is present after RBC transfusions.
 Serum iron and ferritin increase progressively throughout life due to RBC transfusions.
Serum iron and ferritin increase progressively throughout life due to RBC transfusions.
 Serum bilirubin is elevated.
Serum bilirubin is elevated.
 Liver function tests are abnormal, in part the result of transfusional viral hepatitis. This problem is becoming rare with present-day transfusion practice.
Liver function tests are abnormal, in part the result of transfusional viral hepatitis. This problem is becoming rare with present-day transfusion practice.
 LDH and uric acid are elevated.
LDH and uric acid are elevated.
 Haptoglobin is decreased.
Haptoglobin is decreased.
 Endocrine abnormalities are related to extensive iron deposits, with laboratory evidence of hypogonadism and diabetes.
Endocrine abnormalities are related to extensive iron deposits, with laboratory evidence of hypogonadism and diabetes.
 Hypercoagulability: abnormalities in the level of clotting factors and their inhibitors have been reported in some cases.
Hypercoagulability: abnormalities in the level of clotting factors and their inhibitors have been reported in some cases.
β-THALASSEMIA MINOR (TRAIT)
 Definition
Definition
Heterozygotes who carry one normal β-globin allele and one β-thalassemic allele are clinically normal but have an abnormal hematologic picture that may mislead for a diagnosis of iron deficiency.
 Laboratory Findings
Laboratory Findings
 CBC shows microcytic anemia. The anemia is milder (Hg 10–13 g/dL), but the microcytosis is more profound (MCV 60–70 fL) than seen in iron deficiency. RBC count may be higher than normal (another contrast to iron deficiency anemia). RDW is normal, since the RBCs are uniformly microcytic and hypochromic. On peripheral blood smear (PBS), basophilic stippling of RBCs and target cells may be observed. During pregnancy, carriers may develop a more profound anemia, than attributable to the physiologic anemia of pregnancy.
CBC shows microcytic anemia. The anemia is milder (Hg 10–13 g/dL), but the microcytosis is more profound (MCV 60–70 fL) than seen in iron deficiency. RBC count may be higher than normal (another contrast to iron deficiency anemia). RDW is normal, since the RBCs are uniformly microcytic and hypochromic. On peripheral blood smear (PBS), basophilic stippling of RBCs and target cells may be observed. During pregnancy, carriers may develop a more profound anemia, than attributable to the physiologic anemia of pregnancy.
 Hb variant analysis: HbA2 is elevated, sometimes as high as 7–8% with the ratio HbA2/HbA1 being 1:20 instead of the normal 1:40; HgF is slightly elevated in 50% of cases. Some forms of β-thalassemia trait may have a normal concentration of HbA2. Definitive diagnosis can only made by molecular genetic techniques.
Hb variant analysis: HbA2 is elevated, sometimes as high as 7–8% with the ratio HbA2/HbA1 being 1:20 instead of the normal 1:40; HgF is slightly elevated in 50% of cases. Some forms of β-thalassemia trait may have a normal concentration of HbA2. Definitive diagnosis can only made by molecular genetic techniques.
ALPHA-THALASSEMIA SYNDROMES
 Definition
Definition
Normal subjects have four α globin genes, two on each chromosome. The α-thalassemias are caused by mutations or deletions affecting the production of one or more of the four α globin genes. This defect results in a relative excess of β globin chains, which may lead to hemolysis.
 Who Should Be Suspected?
Who Should Be Suspected?
α-Thalassemia should be suspected based on a family history of anemia and geographic and ethnic background. The condition is prevalent in populations of African, Middle East, or Southeast Asian ancestry. The diagnosis is further suspected in cases of microcytic, hypochromic anemia not due to iron deficiency, with normal levels of HbA2 on hemoglobin variant analysis.
 Diagnosis
Diagnosis
The severity of the syndrome depends on the number of α genes affected.
 Loss of all four α globin loci results in hydrops fetalis with Hb Bart, condi tion incompatible with extrauterine life. This condition is not seen in populations of African ancestry, but it is encountered in Asian populations. Hb Bart is composed of four γ globin chains, which fails to deliver oxygen to tissues. It is fast moving on Hb electrophoresis.
Loss of all four α globin loci results in hydrops fetalis with Hb Bart, condi tion incompatible with extrauterine life. This condition is not seen in populations of African ancestry, but it is encountered in Asian populations. Hb Bart is composed of four γ globin chains, which fails to deliver oxygen to tissues. It is fast moving on Hb electrophoresis.
 Loss of three α loci results in Hemoglobin H disease. These patients have a moderate microcytic, hypochromic anemia with inclusion bodies present on the peripheral blood smear (PBS). Hb levels are usually 8–10 g/dL. Hb electrophoresis or chromatographic techniques show 5–30% HbH, which is the result of tetrameric β-chains. HbH disease can be acquired in hematologic malignancies, especially in myelodysplastic syndromes.
Loss of three α loci results in Hemoglobin H disease. These patients have a moderate microcytic, hypochromic anemia with inclusion bodies present on the peripheral blood smear (PBS). Hb levels are usually 8–10 g/dL. Hb electrophoresis or chromatographic techniques show 5–30% HbH, which is the result of tetrameric β-chains. HbH disease can be acquired in hematologic malignancies, especially in myelodysplastic syndromes.
 Loss of two loci results in α-thalassemia-1 trait (a-thalassemia minor). There are two variants, depending if the two affected genes are on the same chromosome, or one per chromosome. Adult patients may have a mild microcytic, hypochromic anemia. In these cases, the red cells are microcytic, hypochromic, and target cells are present. Hb electrophoresis is normal. Definitive diagnosis can only made by molecular genetic techniques.
Loss of two loci results in α-thalassemia-1 trait (a-thalassemia minor). There are two variants, depending if the two affected genes are on the same chromosome, or one per chromosome. Adult patients may have a mild microcytic, hypochromic anemia. In these cases, the red cells are microcytic, hypochromic, and target cells are present. Hb electrophoresis is normal. Definitive diagnosis can only made by molecular genetic techniques.
 Loss of only one locus results in α-thalassemia-2 trait (α-thalassemia minima or silent carrier of α-thalassemia). There are no hematologic abnormalities, and Hb electrophoresis is normal. The diagnosis can only be made by DNA analysis.
Loss of only one locus results in α-thalassemia-2 trait (α-thalassemia minima or silent carrier of α-thalassemia). There are no hematologic abnormalities, and Hb electrophoresis is normal. The diagnosis can only be made by DNA analysis.
 Hemoglobin Constant Spring is a common structural variant associated with α-thalassemia in Asia. It is associated with a normal α chain, but the Constant Spring allele functions as a severe α-thalassemia gene. Patients show a minor, very slowly migrating abnormal Hg component of Hg electrophoresis. Homozygosity results in a mild form of HbH disease.
Hemoglobin Constant Spring is a common structural variant associated with α-thalassemia in Asia. It is associated with a normal α chain, but the Constant Spring allele functions as a severe α-thalassemia gene. Patients show a minor, very slowly migrating abnormal Hg component of Hg electrophoresis. Homozygosity results in a mild form of HbH disease.
 Couples at risk for having offsprings with homozygous thalassemia may choose antenatal diagnosis by direct gene analysis of the fetus.
Couples at risk for having offsprings with homozygous thalassemia may choose antenatal diagnosis by direct gene analysis of the fetus.
Suggested Readings
Benz EJ. Newborn screening for α-thalassemia-keeping up with globalization. N Engl J Med. 2011;364:770–771.
Forget BG. Thalassemia. Hematol Clin North Am. 2010;24:1–140.
Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118:3479–3488.
 HEMOLYTIC INTRINSIC RED BLOOD CELL DEFECTS
HEMOLYTIC INTRINSIC RED BLOOD CELL DEFECTS
ENZYMOPATHIES
The most common enzymopathies are glucose-6-phosphate dehydrogenase (G6PD) and pyruvate kinase (PK) deficiencies. Other rare deficiencies of RBC enzymes do occur but will not be discussed here.
GLUCOSE-6-PHOSPHATE DEHYDROGENASE (G6PD) DEFICIENCY
 Definition
Definition
G6PD deficiency is an X-linked inherited RBC enzyme deficiency. Incidence is high in regions where malaria is or was prevalent. More than 300 variants have been described. G6PD deficiency can be divided into three classes, with the normal genotype being designated G6PD type B.
 Class 1 (Mediterranean variant, also designated G6PD type B−): <5% of normal RBC enzyme activity. It results in a chronic hemolytic anemia exacerbated by oxidant drugs or febrile illnesses. Very severe hemolytic attacks develop after the ingestion of fava beans (favism).
Class 1 (Mediterranean variant, also designated G6PD type B−): <5% of normal RBC enzyme activity. It results in a chronic hemolytic anemia exacerbated by oxidant drugs or febrile illnesses. Very severe hemolytic attacks develop after the ingestion of fava beans (favism).
 Class 2 (African variant, G6PD type A−): <10% of normal RBC enzyme activity; patients have episodic hemolytic attacks produced by certain infections, oxidant drugs, or diabetic ketoacidosis. It is not triggered by ingestion of fava beans.
Class 2 (African variant, G6PD type A−): <10% of normal RBC enzyme activity; patients have episodic hemolytic attacks produced by certain infections, oxidant drugs, or diabetic ketoacidosis. It is not triggered by ingestion of fava beans.
 Class 3: 10–60% of the normal enzyme activity. There is no hemolysis except for limited episodes (2–3 days) after ingestion of oxidant drugs or following infections. Similar G6PD levels are found in female carriers.
Class 3: 10–60% of the normal enzyme activity. There is no hemolysis except for limited episodes (2–3 days) after ingestion of oxidant drugs or following infections. Similar G6PD levels are found in female carriers.
 Who Should Be Suspected?
Who Should Be Suspected?
G6PD deficiency should be considered in the differential diagnosis of a patient with nonimmune (Coombs negative) hemolytic anemia.
 Laboratory Findings
Laboratory Findings
Basis of diagnosis:
 Generation of NADPH from NADP as detected by either quantitative spectrophotometric analysis or, more rapidly, by screening tests, such as a fluorescent spot test.
Generation of NADPH from NADP as detected by either quantitative spectrophotometric analysis or, more rapidly, by screening tests, such as a fluorescent spot test.
 G6PD levels may be normal during and shortly following a hemolytic episode in type A– cases, because very young RBCs contain sufficient enzyme. Assays for G6PD should be postponed for at least 6 weeks after an acute episode.
G6PD levels may be normal during and shortly following a hemolytic episode in type A– cases, because very young RBCs contain sufficient enzyme. Assays for G6PD should be postponed for at least 6 weeks after an acute episode.
CBC: Hemolytic anemia: chronic in class 1 and intermittent in classes 2 and 3. It is seen 2–4 days after ingestion of an oxidant drug (primaquine and sulfa drugs are the most common offending agents) or after fava beans consumption. Female carriers: possible mild hemolytic episodes that are difficult to diagnose with conventional methodology; they can be diagnosed by genetic methods.
 Peripheral blood smear (PBS): Heinz bodies in RBCs (require brilliant cre sylblue supravital special stain), nucleated RBCs, spherocytes, poikilocytes, fragmented RBCs, and bite cells.
Peripheral blood smear (PBS): Heinz bodies in RBCs (require brilliant cre sylblue supravital special stain), nucleated RBCs, spherocytes, poikilocytes, fragmented RBCs, and bite cells.
 Reticulocytosis.
Reticulocytosis.
 MCV may be elevated in type A− patients if not supplemented with folic acid.
MCV may be elevated in type A− patients if not supplemented with folic acid.
 Bilirubin is elevated correlating to the degree of hemolysis. Neonatal jaundice develops in 5% of affected newborns of African or Mediterranean ancestry after the first 24 hours of life. Serum indirect bilirubin reaches a peak (often >20 mg/dL) at 3rd to 5th day with resulting kernicterus if not treated promptly.
Bilirubin is elevated correlating to the degree of hemolysis. Neonatal jaundice develops in 5% of affected newborns of African or Mediterranean ancestry after the first 24 hours of life. Serum indirect bilirubin reaches a peak (often >20 mg/dL) at 3rd to 5th day with resulting kernicterus if not treated promptly.
PYRUVATE KINASE (PK) DEFICIENCY
 Definition
Definition
PK deficiency is an autosomal recessive, nonspherocytic chronic hemolytic anemia. Affected individuals are either homozygous for a single mutation or heterozygous for two different PK mutations. The mechanism of hemolysis has not been elucidated.
 Who Should Be Suspected?
Who Should Be Suspected?
A patient with chronic, sometimes severe, nonimmune (Coombs negative) hemolytic anemia.
There is a wide range of clinical and laboratory findings. The severity of anemia varies from severe neonatal anemia requiring transfusions to a fully compensated hemolytic process in adults who have 10–20% of the normal enzyme in their RBCs. The severely affected patients may require frequent red cell transfusions and as a consequence develop iron storage overload. The severe cases present with jaundice, pallor, and splenomegaly. Such patients may also develop gallstones. The anemia may worsen after certain infections (aplastic crises). PK deficiency is more common in persons of northern European extraction and possibly in Chinese. The disease is particularly severe among the Amish of Pennsylvania.
 Laboratory Findings
Laboratory Findings
 Peripheral blood smear (PBS) shows no characteristic changes, particularly no spherocytes.
Peripheral blood smear (PBS) shows no characteristic changes, particularly no spherocytes.
 The laboratory diagnosis is based on demonstrating low erythrocytic PK enzymatic levels. A screening test using crude hemolysate detects heterozygous carriers in persons who are hematologically normal. This assay may miss some variants. Quantitation of the enzyme can be performed in specialized laboratories.
The laboratory diagnosis is based on demonstrating low erythrocytic PK enzymatic levels. A screening test using crude hemolysate detects heterozygous carriers in persons who are hematologically normal. This assay may miss some variants. Quantitation of the enzyme can be performed in specialized laboratories.
 Genetic tests are the most definitive approach to diagnosis.
Genetic tests are the most definitive approach to diagnosis.
 Elevated levels of LDH and decreased haptoglobin can be seen.
Elevated levels of LDH and decreased haptoglobin can be seen.
HEREDITARY SPHEROCYTOSIS (HS)
 Definition
Definition
HS is a congenital red cell membrane abnormality resulting from defects in one of six genes encoding proteins involved in vertical linkages that tie the membrane skeletal network of the RBCs with the lipid bilayer. The ankyrin gene is the one most commonly involved. HS is inherited as autosomal dominant transmission in 75% of affected individuals. The condition is recessive or presents as a new mutation in the remaining 25%. HS is seen mostly in patients of northern European origin.
 Who Should Be Suspected
Who Should Be Suspected
 Patients with mild to severe anemia, jaundice, splenomegaly, and cholelithiasis early in life, and a family history of a hereditary hemolytic anemia.
Patients with mild to severe anemia, jaundice, splenomegaly, and cholelithiasis early in life, and a family history of a hereditary hemolytic anemia.
 Exacerbations of anemia may occur in aplastic (infections with parvovirus B19 or other viruses), hemolytic crises (with some viral infections), or due to the development of megaloblastic anemia, usually the result of folate deficiency.
Exacerbations of anemia may occur in aplastic (infections with parvovirus B19 or other viruses), hemolytic crises (with some viral infections), or due to the development of megaloblastic anemia, usually the result of folate deficiency.
 Laboratory Findings
Laboratory Findings
 CBC: Anemia of varying severity, but with acute exacerbations (see above). Moderately severe anemia occurs in approximately 70% of cases. Approximately 20% have mild, compensated hemolysis. Approximately 10% of HS patients have severe, debilitating anemia and are transfusion dependent, unless splenectomized (splenectomy ameliorates the anemia, but spherocytosis persists). Indices: normal or slightly low MCV (except elevated when the reticulocyte count is very high or if the patient is folate deficient), elevated MCHC (the most helpful red cell index in HS), and RDW.
CBC: Anemia of varying severity, but with acute exacerbations (see above). Moderately severe anemia occurs in approximately 70% of cases. Approximately 20% have mild, compensated hemolysis. Approximately 10% of HS patients have severe, debilitating anemia and are transfusion dependent, unless splenectomized (splenectomy ameliorates the anemia, but spherocytosis persists). Indices: normal or slightly low MCV (except elevated when the reticulocyte count is very high or if the patient is folate deficient), elevated MCHC (the most helpful red cell index in HS), and RDW.
 Reticulocytosis (5–20%).
Reticulocytosis (5–20%).
 Peripheral blood smear (PBS): Spherocytosis of various degrees is invariably present. Howell-Jolly bodies indicate previous splenectomy. The presence of spherocytes on peripheral blood smear (PBS) is not pathognomonic: it may be due to acquired hemolytic anemias rather than HS.
Peripheral blood smear (PBS): Spherocytosis of various degrees is invariably present. Howell-Jolly bodies indicate previous splenectomy. The presence of spherocytes on peripheral blood smear (PBS) is not pathognomonic: it may be due to acquired hemolytic anemias rather than HS.
 Osmotic fragility reveals increased RBC fragility, but it may be abnormal (increased) also in patients with acquired hemolytic anemias.
Osmotic fragility reveals increased RBC fragility, but it may be abnormal (increased) also in patients with acquired hemolytic anemias.
 Ektacytometry, acidified glycerol lysis test, cryohemolysis test, and especially the flow cytometric eosin-5-maleimide tests have surpassed the osmotic fragility test in sensitivity and specificity but may only be available in specialized laboratories.
Ektacytometry, acidified glycerol lysis test, cryohemolysis test, and especially the flow cytometric eosin-5-maleimide tests have surpassed the osmotic fragility test in sensitivity and specificity but may only be available in specialized laboratories.
 Haptoglobin: decreased.
Haptoglobin: decreased.
 Coombs test: negative.
Coombs test: negative.
 Hemoglobin: usually normal at birth but decreases sharply during the subsequent 20 days of life.
Hemoglobin: usually normal at birth but decreases sharply during the subsequent 20 days of life.
 Bilirubin is slightly elevated, except in neonatal cases with severe hemolysis, when it may be elevated at birth, resulting in kernicterus if not treated promptly.
Bilirubin is slightly elevated, except in neonatal cases with severe hemolysis, when it may be elevated at birth, resulting in kernicterus if not treated promptly.
 Genetic tests: offered in some research laboratories, usually unnecessary.
Genetic tests: offered in some research laboratories, usually unnecessary.
 Other Considerations
Other Considerations
 Laboratory findings may reflect cholelithiasis or aplastic crises.
Laboratory findings may reflect cholelithiasis or aplastic crises.
 Falsely elevated potassium (hyperkalemia) is due to potassium leaking from RBCs.
Falsely elevated potassium (hyperkalemia) is due to potassium leaking from RBCs.
HEREDITARY ELLIPTOCYTOSIS (HE)
 Definition
Definition
HE is a congenital heterogeneous disorder of the membrane skeleton of RBCs, most commonly a defect in spectrin. It is transmitted in an autosomal dominant mode of inheritance. Occasionally, the transmission is recessive. Individuals who are heterozygous for HE are asymptomatic. Individuals who are homozygous or compound heterozygous (10% of patients) exhibit mild to severe anemia. The disorder is more frequent in African Americans and in patients of Mediterranean origin (previous areas of endemic malaria). Affected neonates may have transient overt hemolytic anemia until adult Hb supervenes.
 Laboratory Findings
Laboratory Findings
 Peripheral blood smear (PBS): more than 50% of RBCs are ellipsoidal or rod shaped. Other markers of hemolysis are uncommon, except in the approximately 10% of severely affected patients. In severe cases of HE, severe poikilocytosis is common.
Peripheral blood smear (PBS): more than 50% of RBCs are ellipsoidal or rod shaped. Other markers of hemolysis are uncommon, except in the approximately 10% of severely affected patients. In severe cases of HE, severe poikilocytosis is common.
 Indices: decreased MCV, MCH, MCHC; increased RDW.
Indices: decreased MCV, MCH, MCHC; increased RDW.
 Variant Hb studies and osmotic fragility (see above under HS) are normal.
Variant Hb studies and osmotic fragility (see above under HS) are normal.
 Other Considerations
Other Considerations
 Some degree of elliptocytosis may be seen in peripheral blood smear (PBS) of other types of anemia.
Some degree of elliptocytosis may be seen in peripheral blood smear (PBS) of other types of anemia.
HEREDITARY PYROPOIKILOCYTOSIS (HP)
 Definition
Definition
HP is considered a subtype of HE. In homozygous individuals, it results in a severe congenital hemolytic anemia. It occurs primarily in people of African ancestry.
 Laboratory Findings
Laboratory Findings
Peripheral blood smear (PBS): RBCs are markedly misshapen (fragments, microspherocytes, elliptocytes, pyknotic forms). The RBCs fragment when heated at 45–46°C (normal RBCs show budding and fragmentation only when heated at 49°C). Severe microcytosis and micropoikilocytosis are present.
HEREDITARY OVALOCYTOSIS (HO)
 Definition
Definition
HO is a condition of altered membrane deformability. It is very common in Southeast Asia, where its prevalence is 5–25% in malaria endemic areas. Transmission is autosomal dominant, but so far only heterozygous individuals have been identified. Most affected people express minimal hemolysis.
 Laboratory Findings
Laboratory Findings
Peripheral blood smear (PBS): oval-shaped RBCs with one or two transverse ridges or a longitudinal slit.
 Other Considerations
Other Considerations
Hereditary ovalocytosis can be confused with hereditary elliptocytosis.
HEREDITARY STOMATOCYTOSIS
 Definition
Definition
Hereditary stomatocytosis is an uncommon autosomal dominant disease resulting from defective RBC permeability to sodium and potassium ions.
 Laboratory Findings
Laboratory Findings
Peripheral blood smear (PBS):
 Homozygous individuals: >35% of RBCs show slit-like areas of central pallor, producing a mouth-like appearance.
Homozygous individuals: >35% of RBCs show slit-like areas of central pallor, producing a mouth-like appearance.
 Heterozygous individuals: 1–25% stomatocytes.
Heterozygous individuals: 1–25% stomatocytes.
 Anemia: similar to that of hereditary ovalocytosis.
Anemia: similar to that of hereditary ovalocytosis.
 Homozygous individuals: varying degrees of hemolysis.
Homozygous individuals: varying degrees of hemolysis.
 Heterozygous individuals: no anemia.
Heterozygous individuals: no anemia.
 Other Considerations
Other Considerations
 Stomatocytes may be seen on the peripheral blood smear (PBS) of many acquired disorders, such as alcoholism, liver disease, and drug-induced hemolytic anemias.
Stomatocytes may be seen on the peripheral blood smear (PBS) of many acquired disorders, such as alcoholism, liver disease, and drug-induced hemolytic anemias.
 HEMOLYTIC EXTRINSIC RED BLOOD CELL DEFECTS
HEMOLYTIC EXTRINSIC RED BLOOD CELL DEFECTS
AUTOIMMUNE HEMOLYTIC ANEMIAS (AIHAS)
AIHAs anemias may be classified on the basis of the type of antibody present: warm (binding optimally at 37°C), cold (binding optimally at 4°C), or occasionally combined warm and cold antibodies. Each of these AIHAs may be idiopathic or secondary to other diseases.
 Definition
Definition
Warm-Reactive AIHAs
Warm-reactive AIHAs are due to the presence of IgG antibodies that react with RBC antigens at body temperature. About 60% are idiopathic, and the remaining 40% are the result of lymphomas, leukemias, other neoplasms, or autoimmune disorders such as SLE. They may also accompany HIV or other viral infections.
Cold-Reactive AIHAs and Cold Agglutinin Disease
These AIHAs result from the presence of IgM antibodies that react to polysaccharide antigens on the RBC surface at temperatures below that of the body’s. The antibodies fix complement.
Most of the chronic cases, for which the term cold agglutinin disease is commonly used, have an underlying B-cell neoplasm (CLL/small lymphocytic lymphoma [SLL], lymphomas, macroglobulinemia) as their etiology. Some cases are idiopathic. Acute cases either are the result of viral infections such as mycoplasma pneumonia and infectious mononucleosis or belong to a group known as paroxysmal cold hemoglobinuria. There are variable degrees of hemolysis; the disease can be intravascular or extravascular. The symptoms are exacerbated in cold weather; Raynaud phenomena are common, with vascular obstruction due to RBC clumps, cyanosis of exposed parts, and pallor. Splenomegaly is uncommon; the liver is the site for sequestering the coated RBCs.
 Laboratory Findings
Laboratory Findings
Warm-Reactive AIHA
 Hb moderate to severe decrease, in the range of 7–10 g/dL.
Hb moderate to severe decrease, in the range of 7–10 g/dL.
 Reticulocytes: elevated in most cases.
Reticulocytes: elevated in most cases.
 Indices: increased MCV due to reticulocytosis; increased MCHC reflects the presence of spherocytes.
Indices: increased MCV due to reticulocytosis; increased MCHC reflects the presence of spherocytes.
 Peripheral blood smear (PBS): microspherocytes, polychromasia, and occasionally nucleated RBCs.
Peripheral blood smear (PBS): microspherocytes, polychromasia, and occasionally nucleated RBCs.
 Coombs test: direct IgG and C3d are positive. The warm antibodies are in most cases directed against IgG1 and less frequently against IgG3.
Coombs test: direct IgG and C3d are positive. The warm antibodies are in most cases directed against IgG1 and less frequently against IgG3.
 Unconjugated bilirubin, LDH, urine, and fecal urobilinogen: elevated.
Unconjugated bilirubin, LDH, urine, and fecal urobilinogen: elevated.
 Haptoglobin: decreased.
Haptoglobin: decreased.
Cold-Reactive AIHA and Cold Agglutinin Disease
 Anemia (severity depends on cold agglutinin titer) with anomalous high MCV and MCHC (artifacts due to RBC clumping at room temperature).
Anemia (severity depends on cold agglutinin titer) with anomalous high MCV and MCHC (artifacts due to RBC clumping at room temperature).
 Peripheral blood smear (PBS): RBC clumping.
Peripheral blood smear (PBS): RBC clumping.
 Reticulocyte count: high.
Reticulocyte count: high.
 Anticomplement (C3) Coombs test (positive). Anti-I antibodies are best detected using cord blood red cells.
Anticomplement (C3) Coombs test (positive). Anti-I antibodies are best detected using cord blood red cells.
 Cold agglutinin titers: elevated.
Cold agglutinin titers: elevated.
PAROXYSMAL NOCTURNAL HEMOGLOBINURIA (PNH)
 Definition
Definition
PNH is an acquired disorder of hematopoietic stem cells, characterized by intravascular hemolysis and hemoglobinuria. Only 25% of cases present with classical nocturnal and paroxysmal hemolysis. The clinical triad of intravascular hemolysis, venous thrombosis (the leading cause of death), and bone marrow failure is typical. The chronic intravascular hemolysis results in iron deficiency. There is a high risk of evolution into aplastic anemia, myelodysplastic syndrome, or AML. Clinically, the polymorphism of PNH can be roughly divided into two presentations:
 Classic PNH: hemolysis without bone marrow failure
Classic PNH: hemolysis without bone marrow failure
 Aplastic anemia–PNH syndrome (AA-PNH): hemolysis with bone marrow failure
Aplastic anemia–PNH syndrome (AA-PNH): hemolysis with bone marrow failure
 When To Suspect Paroxysmal Nocturnal Hemoglobinuria
When To Suspect Paroxysmal Nocturnal Hemoglobinuria
 Patients with Coombs-negative intravascular hemolysis, especially if concurrently iron deficient.
Patients with Coombs-negative intravascular hemolysis, especially if concurrently iron deficient.
 Patients with hemoglobinuria.
Patients with hemoglobinuria.
 Patients with venous thrombosis involving unusual sites (mesenteric, hepatic, portal, cerebral, or dermal veins) and especially patients with otherwise unexplained Budd-Chiari syndrome. Such patients should also be investigated for the JAK2 V617F (see p. 1021) mutation if the etiology remains unclear.
Patients with venous thrombosis involving unusual sites (mesenteric, hepatic, portal, cerebral, or dermal veins) and especially patients with otherwise unexplained Budd-Chiari syndrome. Such patients should also be investigated for the JAK2 V617F (see p. 1021) mutation if the etiology remains unclear.
 Patients with unexplained refractory anemia.
Patients with unexplained refractory anemia.
 Laboratory Findings
Laboratory Findings
Highly Recommended
 Flow cytometry (high sensitivity and specificity).
Flow cytometry (high sensitivity and specificity).
 At least two different monoclonal antibodies, directed against two different glycosylphosphatidylinositol (GPI) anchored proteins, either absent or greatly diminished in PNH, on at least two different cell lineages, should be used to diagnose a patient with PNH. In fact, the preferred approach includes the detailed evaluation of leukocytes, because some red blood cells may be lost secondary to hemolysis or diluted from their true frequency by repeated transfusions.
At least two different monoclonal antibodies, directed against two different glycosylphosphatidylinositol (GPI) anchored proteins, either absent or greatly diminished in PNH, on at least two different cell lineages, should be used to diagnose a patient with PNH. In fact, the preferred approach includes the detailed evaluation of leukocytes, because some red blood cells may be lost secondary to hemolysis or diluted from their true frequency by repeated transfusions.
 CD59 and CD55 are the most commonly assessed. Other monoclonal antibodies directed against determinants on leukocytes such as CD14, CD16, and CD24, can be used.
CD59 and CD55 are the most commonly assessed. Other monoclonal antibodies directed against determinants on leukocytes such as CD14, CD16, and CD24, can be used.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


