Stimulatory tests should be performed if hypofunction is suspected and suppression tests if hyperfunction is suspected.
 Suppression tests suppress normal glands but not autonomous secretion.
Suppression tests suppress normal glands but not autonomous secretion.
 Patient preparation is particularly important for hormone studies, results of which may be markedly affected by many factors such as stress, position, fasting state, time of day, preceding diet, and drug therapy. These all should be recorded on the laboratory test requisition form and discussed with the laboratory prior to test ordering.
Patient preparation is particularly important for hormone studies, results of which may be markedly affected by many factors such as stress, position, fasting state, time of day, preceding diet, and drug therapy. These all should be recorded on the laboratory test requisition form and discussed with the laboratory prior to test ordering.
 Appropriate and timely transportation to the laboratory and preparation of specimen are essential.
Appropriate and timely transportation to the laboratory and preparation of specimen are essential.
 No single test adequately reflects the endocrine status in all conditions.
No single test adequately reflects the endocrine status in all conditions.
 Multiple gland hypofunction should evaluate the pituitary gland.
Multiple gland hypofunction should evaluate the pituitary gland.
 DIABETES MELLITUS
DIABETES MELLITUS
 Definition
Definition
The term “diabetes mellitus” (DM) refers to a group of disorders of abnormal carbohydrate metabolism sharing in common the clinical finding of hyperglycemia. DM is associated with a relative or absolute impairment in insulin secretion, along with varying degrees of peripheral resistance to the action of insulin.
 Overview
Overview
DM affects approximately 5% of the world population and 8% of the US population. It is the fourth leading cause of death in the United States. Of the estimated 18 million people with primary DM in the United States, 90–95% have type 2 DM.
 Types and Classification
Types and Classification
The recent classification focuses on the underlying pathophysiologic process, rather than descriptions based upon age at onset or type of treatment.
1.Type 1: immune mediated, results in an absolute insulin deficiency.
2.Type 2: relative insulin deficiency due to abnormalities of both insulin secretion and insulin action. Insulin levels are sufficient to prevent lipid mobilization and ketosis.
3.Gestational diabetes: diagnosed during pregnancy. Only 2% of patients with gestational diabetes remain diabetic after delivery. Forty percent of the patients will develop overt diabetes within 15 years, mostly type 2, but occasionally type 1.
4.Specific types of diabetes:
a.Genetic defects of beta cell function
b.Genetic defects in insulin action
c.Diseases of the exocrine pancreas, such as pancreatitis, trauma, pancreatectomy, neoplasia, cystic fibrosis (CF), hemochromatosis, and fibrocalculous pancreatopathy
5.Associated with endocrinopathies (i.e., Cushing syndrome), drugs (i.e., corticosteroids), or chemicals.
 Who Should Be Suspected?
Who Should Be Suspected?
The clinical onset of diabetes can be acute or insidious, depending both on the degree of insulin deficiency as well as on the intercurrent level of physiologic stress. Patients with the following symptoms and signs should be tested:
1.Classic symptoms of hyperglycemia, such as thirst, polyuria, weight loss, visual blurring
2.Serendipitous finding of hyperglycemia or known impaired glucose tolerance
3.Complications of diabetes, such as proteinuria, neuropathy, cardiovascular complications, and retinopathy
4.Evidence of dehydration, orthostatic hypotension, confusion, or coma
 Screening for Diabetes Mellitus
Screening for Diabetes Mellitus
A.In the absence of specific symptoms
Routine screening for type 1 DM is not recommended, since there is no accepted treatment for the asymptomatic phase of type 1 DM.
However, for type 2 DM, the American Diabetes Association (ADA) recommends screening for diabetes or prediabetes in all adults with body mass index (BMI) ≥25 kg/m2 and one or more additional risk factors for diabetes (see subsequent text of this Chapter). In individuals without risk factors, testing should begin at age 45 years. Fasting plasma glucose is the recommended screening test, since it is faster, easier to perform, more convenient, acceptable to patients, and less expensive.
B.Risk factors for diabetes
1.Age ≥45 years
2.Overweight (body mass index ≥25 kg/m2)
3.Family history diabetes mellitus in a first-degree relative
4.Habitual physical inactivity
5.Belonging to a high-risk ethnic or racial group (e.g., African American, Hispanic, Native American, Asian American, and Pacific Islander)
6.History of delivering a baby weighing >4.1 kg (9 lb) or of gestational DM
7.Hypertension (blood pressure ≥140/90 mm Hg)
8.Dyslipidemia defined as a serum high-density lipoprotein cholesterol concentration ≤35 mg/dL (0.9 mM) and/or a serum triglyceride concentration ≥250 mg/dL (2.8 mM)
9.Previously identified impaired glucose tolerance (IGT) or impaired fasting glucose (IFG)
10.Polycystic ovary syndrome
11.History of vascular disease
 How to Confirm the Diagnosis
How to Confirm the Diagnosis
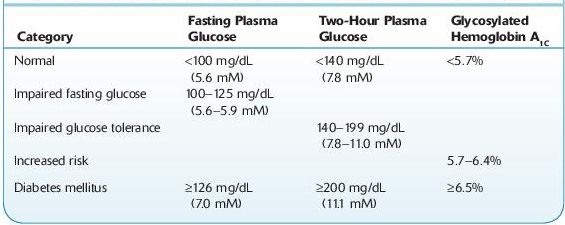
ADA Criteria for the diagnosis of diabetes mellitus:
a.Symptoms of diabetes and a casual plasma glucose ≥200 mg/dL (11.1 mM). Casual is defined as any time of day without regard to time since the last meal. The classic symptoms of diabetes include polyuria, polydipsia, and unexplained weight loss.
Or
b.Fasting plasma glucose ≥126 mg/dL (7.0 mM). Fasting is defined as no caloric intake for at least 8 hours.
Or
c.Two-hour plasma glucose ≥200 mg/dL (11.1 mM) during an oral glucose tolerance test (OGTT). The test should be performed using a glucose load containing the equivalent of 75 g anhydrous glucose dissolved in water.
Or
d.Glycosylated hemoglobin A1c (HbA1c) ≥6.5%. In 2010, the ADA added this as another criterion for the diagnosis of DM. The diagnostic test should be performed using a method that is certified by the National Glycohemoglobin Standardization Program (NGSP) and standardized or traceable to the Diabetes Control and Complications Trial reference assay. Point-of-care HbA1c assays are not sufficiently accurate at this time to use for diagnostic purposes. HbA1c is an extremely valuable clinical tool useful both in the diagnosis and in management of diabetic patients. HbA1c has a circulating life span of about 90 days, and thus the measurement of HbA1c provides information about the level of glycemic control over a 3-month period. However, if the patient’s red blood cells have abnormal survival time, the value of HbA1c may not be reliable. It will be falsely low in patients with hemolytic anemias, and it may be falsely elevated in patients with polycythemia vera or postsplenectomy. It cannot be used as a reliable index for glycemic control in patients with chronic liver diseases due to increased erythrocyte turnover.
In the absence of unequivocal hyperglycemia, the diagnosis of DM must be confirmed on a subsequent day by measuring any one of the three criteria (b, c, and d). However, in symptomatic patients with blood glucose ≥200 mg/dL (11.1 mM) or patients with ketonuria and clear manifestations of type 1 DM, the diagnosis is established and further evaluation is not needed.
Patients with prediabetic conditions (Table 6-1) should be counseled on issues related to lowering their risk for macrovascular diseases (smoking cessation, use of aspirin, diet, and exercise), should have measurements of blood pressure and serum lipids, and should also be encouraged to modify their lifestyle and reduce their weight.
TABLE 6–1. Diagnostic Thresholds for Diabetes and Prediabetic Conditions
 Complications
Complications
Evaluation for complications of diabetes should be done routinely in diabetic patients.
A.Routine eye examination
B.Routine foot examination
C.Screening for microalbuminuria
D.Screening for coronary heart disease
Acute Complications
Excessive and prolonged hyperglycemia associated with uncontrolled diabetes can cause fluid and electrolyte imbalance, which may be life-threatening.
A.Diabetic ketoacidosis (mostly in type 1 DM, may also be seen in type 2 DM): Absolute insulin deficiency leads to the unopposed action of the counterregulatory hormones, including glucagon on the liver, adipose tissue, and muscle, leading to unchecked gluconeogenesis and lipolysis.
a.Signs and symptoms
1.Dehydration, fruity breath smell, orthostatic hypotension, tachypnea, tachycardia, abdominal pain, nausea, vomiting, and confusion
2.Antecedent history of viral or bacterial illness, trauma, or emotional stress
b.Laboratory findings
1.Hyperglycemia (generally ≥300 mg/dL), glucosuria, ketonemia and ketonuria, low bicarbonate, elevated blood urea nitrogen, elevated creatinine, pH usually <7.3.
2.Decreased total body potassium and phosphorus. Serum levels may be normal due to acidosis and shifts to the extracellular space.
B.Hyperosmolar hyperglycemic nonketotic coma: Hyperglycemia in patients with type 2 DM can lead to hyperosmolar coma. The degree of hyperglycemia and dehydration that develop is often far more severe than in patients with type 1 DM.
a.Signs and symptoms
1.Usually occurs in elderly patients with decreased ability to obtain free water; precipitated by illness or drugs
2.Deceased mentation, coma
3.Dehydration
b.Laboratory findings
1.Hyperglycemia (glucose often ≥600 mg/dL)
2.Serum osmolarity often ≥320 mOsm/kg
3.Bicarbonate remains ≥15 mEq/L
4.pH remains ≥7.3
Chronic Complications
A.Microvasculopathy
a.Diabetic nephropathy
1.Diabetes is now the most common cause of end-stage renal disease in Western countries.
2.Twenty to thirty percent of patients with diabetes will develop evidence of nephropathy.
3.The earliest evidence of nephropathy is the appearance of low levels of albumin (30 mg/day or 20 μg/minute) in the urine, termed microalbuminuria.
4.Eighty percent of type 1 DM and 20–40% of type 2 DM patients who develop microalbuminuria will progress to overt nephropathy (≥300 mg/ day or 200 μg/minute) over a period of 10–15 years if not treated.
5.Of those patients who develop overt nephropathy, end-stage renal disease can be expected to develop in 75% of patients with type 1 DM and 20% of patients with type 2 DM over 20 years.
b.Retinopathy and neuropathy
B.Macrovasculopathy and vascular atherosclerosis are also major complications of DM.
Suggested Readings
Khan F, Sachs H, Pechet L. Guide to Diagnostic Testing. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al. Williams Textbook of Endocrinology, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc.; 2008.
Laffel L, Svoren B. Epidemiology, presentation, and diagnosis of type 2 diabetes mellitus in children and adolescents. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Levitsky LL, Misra M. Epidemiology, presentation, and diagnosis of type 1 diabetes mellitus in children and adolescents. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
McCulloch DK. Diagnosis of diabetes mellitus. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
McCulloch DK. Overview of medical care in adults with diabetes mellitus. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
McCulloch DK. Screening for diabetes mellitus. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
 DISORDERS OF THE THYROID GLAND
DISORDERS OF THE THYROID GLAND
THYROTOXICOSIS/HYPERTHYROIDISM
 Definition
Definition
Thyrotoxicosis refers to the classic physiologic manifestations of excessive quantities of the circulating thyroid hormones. The term hyperthyroidism is reserved for disorders that result from sustained overproduction of the hormone by the thyroid itself. Thyrotoxicosis can be caused by hyperthyroidism, or by exogenous thyroid hormone, iatrogenic, or self-administered.
 Overview
Overview
The clinical manifestations of thyrotoxicosis are largely independent of its cause. However, the disorder that causes thyrotoxicosis may have other effects. The most common form is Graves disease, comprising 70–80% of the cases.
 Common Causes
Common Causes
1.Graves disease (diffuse toxic goiter) is the prototypic autoimmune hyperthyroid condition. Prevalence is approximately 1–2% in women; in men, the prevalence is about one tenth of that. Patients commonly have a family history of thyroid dysfunction (hyperthyroidism or hypothyroidism). It may be accompanied by an infiltrative orbitopathy and ophthalmopathy. In patients and their relatives, there is an increased frequency of other autoimmune disorders, such as DM, pernicious anemia, and myasthenia gravis. The radioactive iodine uptake (RAIU) is typically elevated unless the patient has been exposed to excess iodine or acutely to large dose of glucocorticoids. The circulating autoantibodies specific to Graves disease are directed against the thyroid-stimulating hormone (TSH) receptor and can be measured directly.
2.Toxic multinodular goiter (MNG) is a disorder in which hyperthyroidism arises in a multinodular goiter, usually of long standing. The overproduction of thyroid hormone is usually less than in Graves disease and is almost never accompanied by infiltrative ophthalmopathy. All patients with MNG should be screened annually with a serum TSH.
3.Toxic adenoma is usually caused by a single adenoma sometimes referred to as hyperfunctioning solitary nodule or toxic nodule. It often shows a suppressed TSH, which appears in a radioiodine thyroid scan as a localized area of increased radioiodine accumulation.
4.Chorionic gonadotropin–induced hyperthyroidism can be physiologic during pregnancy (transient gestational thyrotoxicosis) or associated with trophoblastic tumors.
5.Iodide-induced hyperthyroidism. Administration of supplemental iodine to subjects with endemic iodine deficiency goiter can result in iodide-induced hyperthyroidism. Amiodarone, an antiarrhythmic medication, is the most common drug that has been reported to be associated with iodine-induced thyrotoxicosis.
6.Autoimmune (Hashimoto’s) thyroiditis can be associated with transient thyrotoxicosis, which is caused by thyroid cell breakdown, and the hyperthyroid symptoms are of abrupt onset and short duration.
7.Subacute thyroiditis is an acute inflammatory disorder of the thyroid gland, which is caused directly or indirectly by a viral infection. The symptoms of fever, malaise, and neck soreness frequently overshadow the symptoms of hyperthyroidism. Characteristic findings are of a tender thyroid gland, an elevated erythrocyte sedimentation rate (ESR), and a low RAIU.
8.Excess thyroid hormone ingestion can be either iatrogenic or factitious. The presence of a low, rather than elevated, serum thyroglobulin level in a patient with thyrotoxic manifestations and a low RAIU is very suspicious for exogenous hormone ingestion rather than thyroid hyperfunction.
9.Thyroid storm (accelerated hyperthyroidism) represents an extreme accentuation of thyrotoxicosis. It is an uncommon but serious complication, with a mortality of 10–75%. Manifestations include severe fever, marked tachycardia, cardiac arrhythmias, tremulousness, and altered mental status.
10.Subclinical (mild) hyperthyroidism refers to the situation that there are no signs or symptoms of thyrotoxicosis, but the serum TSH is subnormal despite normal serum free thyroid hormone concentrations. The diagnosis requires several subnormal TSH concentration results spaced months apart.
11.Ectopic thyroid hormone excretion from the ovary (struma ovarii).
 Who Should Be Suspected?
Who Should Be Suspected?
Signs and symptoms of thyrotoxicosis include
1.Anxiety, emotional lability, nervousness, and irritability
2.Heat intolerance and increased perspiration
3.Weight loss despite a normal or increased appetite
4.Tremor, palpitations, tachycardia, proximal muscle weakness, and exophthalmos
5.Oligomenorrhea in women; gynecomastia and erectile dysfunction in men
 Laboratory Findings
Laboratory Findings
The availability of sensitive and reliable assays for serum TSH and free thyroxine (T4) has made the laboratory diagnosis of hyperthyroidism rather straightforward (Figure 6-1).
 Serum TSH is the most cost-effective screening test. If the value is normal, the patient is very unlikely to have hyperthyroidism. In hyperthyroidism, serum TSH is below normal and frequently <0.1 μIU/mL. TSH may remain decreased for many months in treated formerly hyperthyroid patients; therefore, thyroid hormone levels more accurately reflect the clinical situation.
Serum TSH is the most cost-effective screening test. If the value is normal, the patient is very unlikely to have hyperthyroidism. In hyperthyroidism, serum TSH is below normal and frequently <0.1 μIU/mL. TSH may remain decreased for many months in treated formerly hyperthyroid patients; therefore, thyroid hormone levels more accurately reflect the clinical situation.
 Serum free T4 is important to confirm and determine the degree of hyperthyroidism in a patient with a low TSH.
Serum free T4 is important to confirm and determine the degree of hyperthyroidism in a patient with a low TSH.
 Serum T3 is usually elevated with hyperthyroidism. Assessment of T3 levels is important to determine the severity of the hyperthyroidism and to monitor the response to treatment.
Serum T3 is usually elevated with hyperthyroidism. Assessment of T3 levels is important to determine the severity of the hyperthyroidism and to monitor the response to treatment.
 RAIU is often elevated in Graves disease. However, the diagnostic accuracy of RAIU in hyperthyroidism does not approach that of the serum TSH plus free T4 measurement. Therefore, determining RAIU is not useful in the diagnosis of straightforward Graves disease but is useful in excluding thyrotoxicosis not caused by hyperthyroidism. Very low values of RAIU in association with thyrotoxicosis signal the presence of factitious thyrotoxicosis, ectopic thyroid tissue, subacute thyroiditis, or the thyrotoxic phase of autoimmune thyroiditis.
RAIU is often elevated in Graves disease. However, the diagnostic accuracy of RAIU in hyperthyroidism does not approach that of the serum TSH plus free T4 measurement. Therefore, determining RAIU is not useful in the diagnosis of straightforward Graves disease but is useful in excluding thyrotoxicosis not caused by hyperthyroidism. Very low values of RAIU in association with thyrotoxicosis signal the presence of factitious thyrotoxicosis, ectopic thyroid tissue, subacute thyroiditis, or the thyrotoxic phase of autoimmune thyroiditis.
 Thyrotropin receptor autoantibodies are present in 70–100% of the patients with Graves disease, and their measurement is not usually necessary for diagnosis, but it may be helpful in prognosis because patients who have high titers that do not decrease with antithyroid drug treatment are unlikely to go into remission. Measurement of thyrotropin receptor autoantibodies is important in pregnancy, because a high titer at the end of pregnancy correlates with an increased risk of neonatal hyperthyroidism.
Thyrotropin receptor autoantibodies are present in 70–100% of the patients with Graves disease, and their measurement is not usually necessary for diagnosis, but it may be helpful in prognosis because patients who have high titers that do not decrease with antithyroid drug treatment are unlikely to go into remission. Measurement of thyrotropin receptor autoantibodies is important in pregnancy, because a high titer at the end of pregnancy correlates with an increased risk of neonatal hyperthyroidism.
 Abnormal TSH can also been seen in various nonthyroidal diseases. Simultaneous measurement of TSH with free T4 is useful in evaluating the differential diagnoses.
Abnormal TSH can also been seen in various nonthyroidal diseases. Simultaneous measurement of TSH with free T4 is useful in evaluating the differential diagnoses.
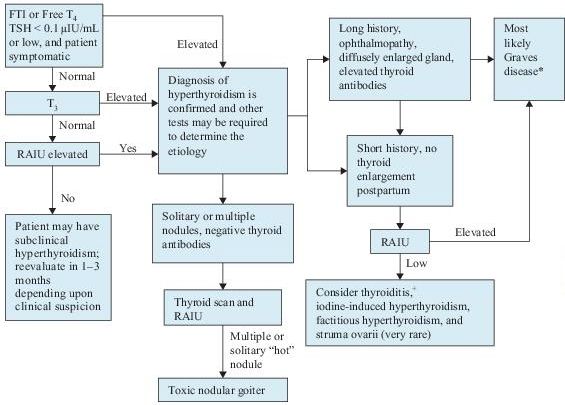
Figure 6–1 Algorithm for the diagnosis of hyperthyroidism. *Graves disease can be confirmed by measuring thyroid antibodies. +Suspect postpartum thyroiditis if within 6 months of delivery, subacute thyroid if associated with tender gland and constitutional symptoms, and silent thyroiditis if neither. T4 , thyroxine; FTI, free thyroxine index; RAIU, radioactive iodine uptake; T3 , triiodothyronine; TSH, thyroidstimulating hormone.
Suggested Readings
Khan F, Sachs H, Pechet L, et al. Guide to Diagnostic Testing. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al. Williams Textbook of Endocrinology, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc., 2008.
Ross DS. Diagnosis of hyperthyroidism. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Ross DS. Overview of the clinical manifestations of hyperthyroidism in adults. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
HYPOTHYROIDISM
 Definition
Definition
Hypothyroidism refers to a condition in which the amount of thyroid hormones in the body is below normal.
 Overview
Overview
The diagnosis of hypothyroidism relies heavily upon laboratory tests because of the lack of specificity of the typical clinical manifestations. The prevalence of hypothyroidism is approximately 5% in adults and 15% in women older than 65 years of age. Hypothyroidism is less common in men, with a five to eight times lower incidence. Hypothyroidism is far more common than hyperthyroidism. Hypothyroidism is usually easily treated with thyroid hormone replacement. It is now hypothesized that autoimmune hyperthyroidism (Graves disease) and hypothyroidism (Hashimoto thyroiditis) represent two extremes of one spectrum of autoimmune thyroid disease.
 Common Causes
Common Causes
I.Primary hypothyroidism
A.Hashimoto thyroiditis is the most common cause of hypothyroidism in areas of the world in which dietary iodine is sufficient. It usually presents with goiter, hypothyroidism, or both. Goiter usually develops gradually. The diagnosis of Hashimoto thyroiditis is confirmed by the presence of thyroid autoantibodies, including thyroid peroxidase (TPO) antibody and thyroglobulin antibody.
B.Iatrogenic: Thyroidectomy and radioiodine therapy or external irradiation for the treatment of carcinoma, hyperthyroidism, or goiter can lead to hypothyroidism.
C.Iodine deficiency (endemic goiter) almost always occurs in areas of environmental iodine deficiency. The incidence of endemic goiter has been greatly reduced by the introduction of iodized salt.
D.Drugs: thioamides, lithium, amiodarone, interferon, and interleukin-2
E.Infiltrative diseases such as fibrous thyroiditis, hemochromatosis, and sarcoidosis
F.Transient hypothyroidism is defined as a period of reduced free T4 with suppressed, normal, or elevated TSH levels that are eventually followed by an euthyroid state. This form of hypothyroidism usually occurs in the clinical context of subacute (postviral) thyroiditis, lymphocytic (painless) thyroiditis, or postpartum thyroiditis
G.Congenital thyroid agenesis, dysgenesis, or defect in hormone synthesis
H.Subclinical hypothyroidism is defined as a normal serum free T4 concentration and a slightly high serum TSH concentration. These patients usually have nonspecific symptoms and a substantial proportion of them eventually develop overt hypothyroidism
II.Secondary and tertiary (central) hypothyroidism refers to hypothyroidism induced by deficiency of either TSH or thyrotropin-releasing hormone (TRH). This type of hypothyroidism is much less common than primary hypothyroidism, and the symptoms are usually milder than in primary hypothyroidism.
III.Generalized thyroid hormone resistance
 Who Should Be Suspected?
Who Should Be Suspected?
Signs and symptoms of hypothyroidism include:
1.Fatigue, weight gain, depression, and cold intolerance
2.Dry skin, brittle hair, constipation, and muscle cramps
3.Hypermenorrhea in women
4.Thyroid enlargement (goiter), puffy face and hands (myxedema), and delayed ankle reflex relaxation phase
5.Hypothyroidism in infants and children leads to retardation of mental development and of growth. Severe hypothyroidism in infancy is termed cretinism.
6.Myxedema coma refers to severe prolonged hypothyroidism, which is manifested by bradycardia, congestive heart failure, hypothermia, hypoventilation, and paralytic ileus. It is an uncommon but life-threatening condition if not detected and treated promptly.
7.Secondary and tertiary hypothyroidism should be suspected in patients with known hypothalamic or pituitary disease, patients with a pituitary mass, or in patients with other hormonal deficiencies.
 Laboratory Findings (Figure 6-2)
Laboratory Findings (Figure 6-2)
 Laboratory confirmation of the diagnosis of hypothyroidism consists of measuring serum TSH and free T4 . Primary hypothyroidism is characterized by a high serum TSH concentration and a low serum free T4 concentration. Secondary hypothyroidism is characterized by a low serum TSH concentration as well as a low serum T4 concentration.
Laboratory confirmation of the diagnosis of hypothyroidism consists of measuring serum TSH and free T4 . Primary hypothyroidism is characterized by a high serum TSH concentration and a low serum free T4 concentration. Secondary hypothyroidism is characterized by a low serum TSH concentration as well as a low serum T4 concentration.
 Total T4 , RAIU, and free T4 index are usually decreased in hypothyroidism, but they are less sensitive than TSH and free T4 measurement.
Total T4 , RAIU, and free T4 index are usually decreased in hypothyroidism, but they are less sensitive than TSH and free T4 measurement.
 Antithyroid peroxidase (TPO) antibodies are detected in almost all patients with Hashimoto disease and its variants, in 70% of patients with Graves disease, and in a smaller number of patients with various other thyroid disorders such as MNG, nontoxic goiter, and thyroid carcinoma.
Antithyroid peroxidase (TPO) antibodies are detected in almost all patients with Hashimoto disease and its variants, in 70% of patients with Graves disease, and in a smaller number of patients with various other thyroid disorders such as MNG, nontoxic goiter, and thyroid carcinoma.
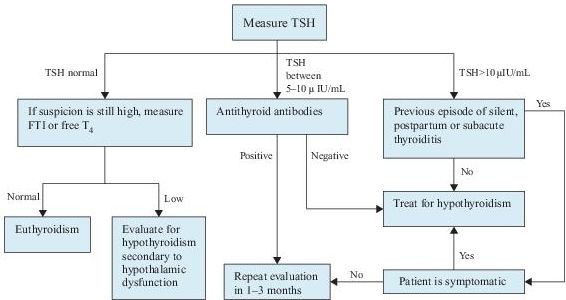
Figure 6–2 Algorithm for the diagnosis of hypothyroidism. T4 , thyroxine; FTI, free thyroxine index; TSH, thyroid-stimulating hormone.
Suggested Readings
Khan F, Sachs H, Pechet L, et al. Guide to Diagnostic Testing. Philadelphia, PA: Lippincott Williams & Wilkins, 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al. Williams Textbook of Endocrinology, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc., 2008.
Ross DS. Diagnosis of and screening for hypothyroidism. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Ross DS. Subclinical hypothyroidism. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
GOITER AND THYROID NODULES
 Definition
Definition
Goiter refers to an enlargement of the thyroid gland. It can be classified in different ways. Toxic goiter refers to goiter with hyperthyroidism. Nontoxic goiter refers to an enlarged thyroid gland with normal or low thyroid hormone levels.
A thyroid nodule is defined as a discrete lesion within the thyroid gland that is due to an abnormal focal growth of thyroid cells.
 Overview
Overview
Thyroid enlargement or nodules come to clinical attention when noted by the patient, or as an incidental finding during routine physical examination, or during a radiologic procedure, such as carotid ultrasonography or neck computed tomography (CT).
The prevalence of goiter, diffuse or nodular, differs widely depending on the iodine intake by the population living in a given area. In the general population, prevalence of 4.6% has been reported as being clinically detected. By using ultrasound as a screening method, a prevalence of up to 30–50% of an unselected adult population has been described as having goiter.
The clinical importance of thyroid nodules is related primarily to the need to exclude thyroid cancer, which accounts for 4–6.5% of all thyroid nodules in non-surgical series. The diagnostic goal is to efficiently identify those patients who require surgical intervention. A solitary nodule should be evaluated for malignancy no matter what the underlying thyroid disorder is.
 Common Causes
Common Causes
I.Diffuse enlargement of the thyroid gland is seen in the following conditions:
 Diffuse toxic goiter—Graves disease; most common cause of endogenous hyperthyroidism
Diffuse toxic goiter—Graves disease; most common cause of endogenous hyperthyroidism
 Diffuse nontoxic (simple) goiter—relative deficiency of thyroid hormone
Diffuse nontoxic (simple) goiter—relative deficiency of thyroid hormone
 Hashimoto thyroiditis
Hashimoto thyroiditis
 Organification defect (abnormality in the incorporation of iodine into thyroid hormone precursors)
Organification defect (abnormality in the incorporation of iodine into thyroid hormone precursors)
II.Nodular enlargement of the thyroid gland is seen in the following situations:
A.Benign solid nodule.
 Hyperplastic (or colloid) nodule
Hyperplastic (or colloid) nodule
 Follicular adenoma
Follicular adenoma
B.Malignant tumors.
 Thyroid carcinomas, including papillary, follicular, anaplastic, and medullary carcinomas
Thyroid carcinomas, including papillary, follicular, anaplastic, and medullary carcinomas
Papillary/follicular/anaplastic carcinomas arise from thyroid follicular epithelial cells. Papillary and follicular cancers are considered differentiated cancers, and patients with these tumors are often treated similarly despite numerous biologic differences. Most anaplastic (undifferentiated) cancers appear to arise from differentiated cancers.
Medullary carcinoma arises from calcitonin-secreting C cells and can occur in both sporadic and hereditary forms. The sporadic (noninherited) form accounts for 80% of cases and is usually unilateral. The hereditary form makes up 20% of the cases, is usually multicentric, and can be transmitted as a single entity and part of multiple endocrine neoplasia (MEN) types 2A and 2B, and familial non-MEN.
 Lymphomas. Most primary thyroid lymphomas arise in patients who have chronic autoimmune thyroiditis.
Lymphomas. Most primary thyroid lymphomas arise in patients who have chronic autoimmune thyroiditis.
C.Multinodular goiter can present with or without thyrotoxicosis. A retrospective study showed that the risk of malignancy was similar in patients with multinodular goiter and one or more dominant nodules to the patients with solitary nodule. Therefore, a dominant nodule in a multinodular goiter should be evaluated as if it were a single nodule.
D.Simple cyst.
 Who Should Be Suspected?
Who Should Be Suspected?
As mentioned earlier, thyroid nodules can be noted by the patient on self-examinations or by the physician on routine physical examinations. In addition, the presence of goiter or thyroid nodules should be suspected in patients with the following symptoms or signs.
1.Pain, pressure, or fullness in the neck
2.Hoarseness or change in voice
3.Trouble swallowing
 Laboratory Findings (Figure 6-3)
Laboratory Findings (Figure 6-3)
1.Serum TSH should be measured in any patient with a goiter or nodules. It may be used as a first-line screening test. In multinodular goiter, TSH usually is in normal or low-normal range; it is rarely increased.
2.Calcitonin level is increased in virtually all patients with clinical medullary carcinoma. However, it is not cost-effective or necessary in patients without clinical suspicion due to rarity of the disease and high frequency of false-positive results.
3.Measurement of serum antithyroid peroxidase antibody and antithyroglobulin antibody levels may be helpful in the diagnosis of chronic autoimmune thyroiditis, especially if the serum TSH level is elevated.
4.Fine needle aspiration (FNA) biopsy of the nodule is the most time- and cost-efficient evaluation. The reported overall rates of sensitivity and specificity exceed 90% in iodine-sufficient geographic areas. FNA biopsy should be performed in any patient with a solitary or predominant nodule in a multinodular gland, unless the TSH is suppressed, implying autonomous function and, therefore, a low likelihood of malignancy.
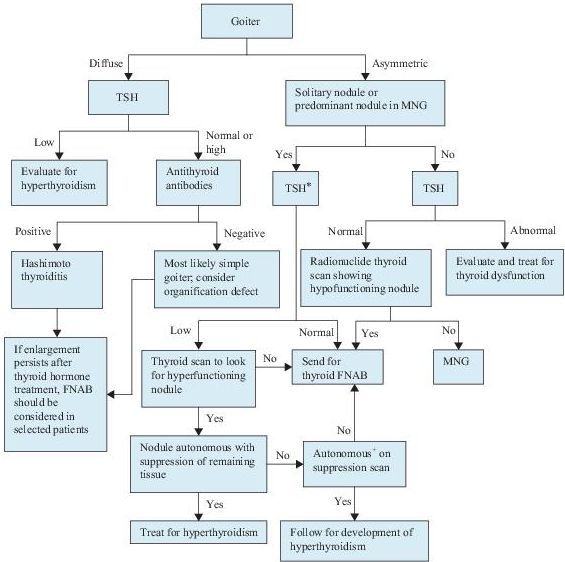
Figure 6–3 Algorithm for the diagnosis of goiter and thyroid nodules. *Include measurement of serum calcitonin if there is a family history of medullary cancer or multiple endocrine neoplasm, type 2 (MEN2). + Autonomy is defined as the ability to concentrate radioactive iodine despite TSH suppression. FNAB, fine needle aspiration biopsy; MNG, multinodular goiter; TSH, thyroid-stimulating hormone.
 Imaging Studies (see Figure 6-3)
Imaging Studies (see Figure 6-3)
1.Ultrasonography should be used to assess both morphology and size of the goiter and assist in screening and follow-up of thyroid nodules that are difficult to palpate. It may also be useful in directing a FNA biopsy in selected patients. However, this technique cannot distinguish between benign and malignant nodules.
2.Thyroid scintigraphy. Radionuclide scans can be performed with either iodine-123 or technetium-99m pertechnetate. Most thyroid carcinomas are inefficient in trapping and organifying iodine and appear as cold nodules. Unfortunately, most benign nodules also do not concentrate iodine and, therefore, are cold nodules. The only situation in which an iodine scan can exclude malignancy with reasonable certainty is in the case of a toxic adenoma, which is characterized by significantly increased uptake within the nodule, so-called “hot” nodule, and markedly suppressed or absent uptake in the remainder of the gland.
Suggested Readings
Khan F, Sachs H, Pechet L, et al. Guide to Diagnostic Testing. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al. Williams Textbook of Endocrinology, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc.; 2008.
Ross DS. Clinical manifestations and evaluation of obstructive or substernal goiter. Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Ross DS. Diagnostic approach to and treatment of thyroid nodules. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
 DISORDERS OF THE ADRENAL GLAND
DISORDERS OF THE ADRENAL GLAND
CUSHING SYNDROME
 Definition
Definition
Cushing syndrome refers to hypercortisolism of any cause. Whereas, Cushing disease refers to hypercortisolism due to an adrenocorticotropic hormone (ACTH)-producing pituitary adenoma.
 Overview
Overview
The incidence of Cushing disease is 5–25 cases per 1,000,000 people per year. Other causes of Cushing syndrome are much less common.
 Common Causes
Common Causes
Cushing syndrome may be either ACTH dependent or ACTH independent.
I.ACTH-dependent Cushing syndrome
A.Cushing disease is the most common cause of Cushing syndrome and comprises 65–70% of the cases. Almost all patients with Cushing disease have a pituitary adenoma. The adenomas are frequently small, and even a gadolinium-enhanced, high-resolution magnetic resonance imaging (MRI) of the sella identifies only 50% of them. Pituitary adenoma cells have a higher than normal set point for cortisol feedback inhibition. This feature is clinically important because it permits the use of dexamethasone suppression to distinguish between pituitary and ectopic ACTH secretion; the latter is usually very resistant to glucocorticoid negative feedback.
B.Ectopic ACTH secretion by nonpituitary tumors accounts for 10–15% of the cases of Cushing syndrome. A wide variety of tumors, usually carcinomas rather than sarcomas or lymphomas, have been associated with ectopic ACTH secretion. The most common causes are small cell carcinomas of the lung, bronchial or pulmonary carcinoid tumors, and pancreatic islet cell tumors and thymic tumors. Ectopic secretion of ACTH causes bilateral adrenocortical hyperplasia and hyperfunction.
C.Ectopic corticotropin-releasing hormone (CRH) syndrome constitutes <1% of Cushing syndrome. CRH secretion by nonhypothalamic tumors causes pituitary hyperplasia, hypersecretion of ACTH, and bilateral adrenal hyperplasia.
II.ACTH-independent Cushing syndrome
A.Adrenal tumors account for 18–20% of the cases of Cushing syndrome. It is important to be sure of the biochemical diagnosis prior to performing any adrenal imaging, since 4% of patients have an adrenal incidentaloma.
B.Iatrogenic or factitious Cushing syndrome is usually caused by the use of prednisone, or potent inhaled, injected, and topical glucocorticoids, such as beclomethasone and fluocinolone. Exogenous glucocorticoids inhibit CRH and ACTH secretion, leading to bilateral adrenocortical atrophy. Plasma ACTH, serum cortisol, and urinary cortisol excretion are all low.
 Who Should Be Suspected?
Who Should Be Suspected?
Symptoms and signs of Cushing syndrome include hypertension, type 2 DM, and menstrual and psychiatric disorders. Physical examination findings include central obesity, proximal muscle weakness, wide purple striae, spontaneous ecchymoses, and facial plethora (moon face).
 Laboratory Findings
Laboratory Findings
I.Diagnosis of Cushing syndrome involves three steps (Figure 6-4). The first step is to suspect Cushing syndrome based on the symptoms and signs. The second step is to confirm the presence of excess cortisol production by biochemical testing. The third step is to determine if the hypercortisolism is ACTH dependent, and, if so, the source of the ACTH.
II.Tests used to establish the diagnosis of Cushing syndrome are listed in Table 6-2. Urinary cortisol, late night salivary cortisol, and low-dose dexamethasone suppression tests are now recommended as first-line tests. At least two first-line tests should be unequivocally abnormal to establish the diagnosis of Cushing syndrome. Urinary and salivary cortisol measurements should be obtained at least twice.
A.Twenty-four–hour urinary cortisol excretion provides a direct and reliable practical index of cortisol secretion. It is an integrated measurement of plasma free cortisol; as cortisol secretion increases, the binding capacity of cortisol-binding globulin is exceeded and results in a disproportionate rise in urinary free cortisol. The two most important factors in obtaining a valid result are collection of a complete 24-hour specimen and a reliable reference laboratory.
B.Late-night or midnight salivary cortisol concentration can also be used. Saliva is easily collected, and cortisol is stable in saliva for several days even at room temperature. The criteria used to interpret salivary cortisol results vary among different studies. Midnight salivary cortisol is an accurate diagnostic test. A cortisol value >2.0 ng/mL has 100% sensitivity and 96% specificity for diagnosing Cushing syndrome.
C.Low-dose dexamethasone suppression tests include an overnight 1-mg test and a standard 2-day test. In normal patients, the administration of glucocorticoid results in suppression of ACTH and cortisol secretion. Whereas in Cushing syndrome of whatever cause, there is a failure of this suppression and the cortisol concentration remains elevated.
D.Midnight serum cortisol is based on the fact that the normal evening or night nadir in serum cortisol is preserved in obese and depressed patients (pseudo-Cushing syndrome) but not in those with Cushing syndrome. The test needs to be repeated on at least two nights. Accuracy of midnight cortisol requires an indwelling catheter, and it is clearly not convenient in an outpatient setting.
III.Tests used to localize the source of the hormone excess: Once the diagnosis of Cushing syndrome is confirmed, the next step is to distinguish among the three most common causes: a pituitary tumor, ectopic ACTH secretion, and an adrenal tumor. Determining whether elevated cortisol is ACTH dependent (due to an ACTH-secreting tumor) or whether it is ACTH independent (due to a primary adrenal disorder) is based primarily on measuring plasma ACTH level.
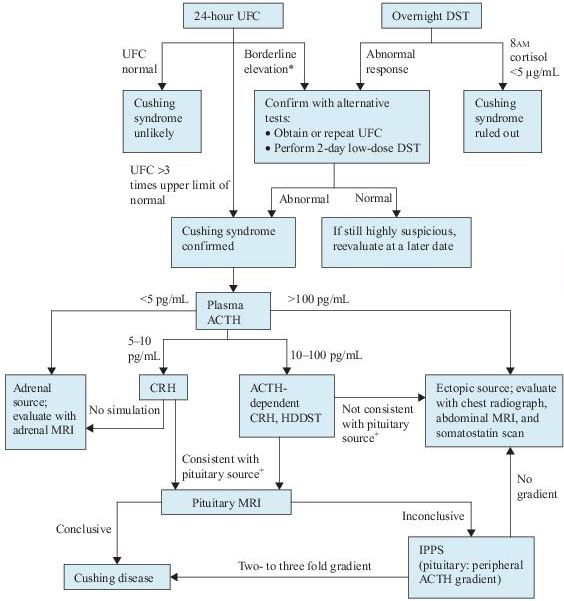
Figure 6–4 Algorithm for the evaluation of Cushing syndrome. *Patients with alcoholism or depression may have pseudo-Cushing syndrome and require a CRH test for further evaluation. +With a pituitary source, ACTH should increase with CRH, and cortisol production should decrease with HDDST. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; DST, dexamethasone suppression test; HDDST, high-dose dexamethasone suppression test; MRI, magnetic resonance imaging; IPPS, inferior petrosal sinus sampling; UFC, urinary free cortisol.
TABLE 6–2. Common Tests Used to Establish the Diagnosis of Cushing Syndrome
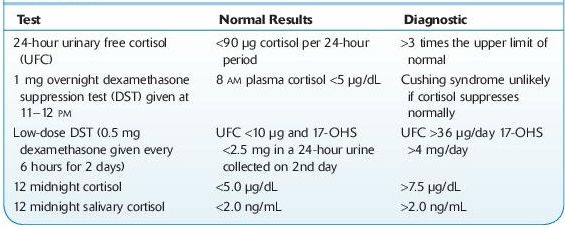
17-OHS, 17-hydroxycorticosteroid.
 Imaging Studies (see Figure 6-4)
Imaging Studies (see Figure 6-4)
1.Adrenal imaging is indicated when plasma ACTH levels are <5 pg/mL. Thinsection CT or MRI is the next step in evaluating the adrenals. Bilateral adrenal hyperplasia may be present in ACTH-dependent disease.
2.Somatostatin scanning. Ectopic sources of ACTH are notoriously difficult to identify. Because many of these tumors are carcinoids and have somatostatin receptors, scintigraphy with the somatostatin analog indium-111-pentreotide can sometimes localize tumors not found by conventional techniques.
3.Because both incidental pituitary and adrenal tumors are common, biochemical evaluation should be completed before any imaging studies.
 Additional Study
Additional Study
Petrosal sinus sampling is used when the anatomic localization fails to identify an unequivocal lesion as suggested by the biochemical testing. This test allows confirmation of the pituitary source of ACTH and identifies the side of the ACTHsecreting lesion. ACTH is measured simultaneously in samples from catheters placed in the left and right inferior petrosal sinuses and compared to peripheral levels. A gradient of two- to threefold is consistent with a pituitary source of ACTH. CRH can also be given during the procedure to enhance its accuracy.
Suggested Readings
Khan F, Sachs H, Pechet L, et al. Guide to Diagnostic Testing. Philadelphia, PA: Lippincott Williams & Wilkins; 2002.
Kronenberg HM, Melmed S, Polonsky KS, et al. Williams Textbook of Endocrinology, 11th ed. Philadelphia, PA: Saunders, Elsevier Inc.; 2008.
Nieman LK. Causes and pathophysiology of Cushing’s syndrome. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Nieman LK. Clinical manifestations of Cushing’s syndrome. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Nieman LK. Establishing the cause of Cushing’s syndrome. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
Nieman LK. Establishing the diagnosis of Cushing’s syndrome. In: Rose B, (ed). UpToDate, Waltham, MA: UpToDate, Inc.; 2009.
ADRENAL INSUFFICIENCY
 Definition
Definition
Adrenal insufficiency is defined as a deficiency of hormones synthesized by the adrenal cortex.
 Common Causes
Common Causes
I.Primary adrenal insufficiency (Addison disease): due to intrinsic diseases of the adrenal glands
A.Autoimmune adrenalitis. It is the most common cause of primary adrenal insufficiency and comprises approximately 70–80% of the cases. Some of the patients also have other autoimmune disorders, such as hypoparathyroidism, type 1 DM, Hashimoto thyroiditis, Graves disease, or pernicious anemia.
B.Infections. Common infectious etiologies include tuberculosis, fungi (histoplasmosis, paracoccidioidomycosis), bacteria (meningococcemia, Pseudomonas aeruginosa), and viruses (HIV, CMV).
C.Adrenal hemorrhage or infarction. Adrenal hemorrhage has been associated with meningococcemia (Waterhouse-Friderichsen syndrome) or Pseudomonas aeruginosa. Anticoagulants are a major risk factor for adrenal hemorrhage.
D.Metastatic disease. Infiltration of the adrenal glands by metastatic cancers is common. The primary site includes the lung, breast, stomach, and colon. Similar findings can be seen with melanomas or lymphomas.
E.Drugs. Several drugs may cause adrenal insufficiency by inhibiting cortisol biosynthesis. They include etomidate, ketoconazole, metyrapone, and suramin.
F.Other risk factors include antiphospholipid syndrome, thromboembolic disease, trauma, stress, adrenoleukodystrophy, and abetalipoproteinemia.
II.Secondary adrenal insufficiency: due to inadequate ACTH secretion by the pituitary
A.Panhypopituitarism. Symptoms are due to a decrease in all pituitary hormones, resulting in hypoadrenalism.
B.Isolated ACTH deficiency.
C.Megestrol acetate. Megestrol is used as an appetite stimulant in patients with metastatic breast cancer or AIDS. It suppresses the hypothalamic–pituitary–adrenal axis.
III.Tertiary adrenal insufficiency: due to inadequate CRH secretion by the hypothalamus
A.Following abrupt cessation of high-dose glucocorticoid therapy
B.Following correction of Cushing syndrome
 Who Should Be Suspected?
Who Should Be Suspected?
Clinical symptoms and signs of adrenal insufficiency vary depending on the rate and extent of loss of adrenal function, whether mineralocorticoid production is preserved, and the degree of stress.
1.Adrenal crisis. It refers to acute adrenal insufficiency, and the predominant manifestation is shock. Other symptoms include anorexia, nausea, vomiting, abdominal pain, weakness, fatigue, lethargy, confusion, or coma. Adrenal crisis may occur in patients with gradual onset who have been stressed by infection, trauma, or surgery.
2.The most common symptoms of chronic adrenal insufficiency are chronic malaise, anorexia, nausea, vomiting, and generalized weakness.
3.Patients with long-standing primary adrenal insufficiency may present with hyperpigmentation. Other frequent signs are hypotension or orthostatic hypotension. Calcification of the auricular cartilage occurs exclusively in men.
4.Patients with secondary and tertiary adrenal insufficiency usually have intact mineralocorticoid function and do not develop hyponatremia and/or hyperkalemia.
 Laboratory Findings (Figure 6-5)
Laboratory Findings (Figure 6-5)
1.Serum cortisol concentration. Cortisol is secreted in a diurnal pattern with highest levels in the morning. Levels measured later in the day are unreliable. Healthy people have early morning serum cortisol concentration of >15 μg/dL. Values <15 μg/dL are suggestive of adrenal insufficiency and require further testing.
2.Basal plasma ACTH concentration. An elevated morning ACTH plasma level in the presence of low cortisol is diagnostic of primary adrenal insufficiency. In contrast, plasma ACTH concentrations are low or low normal in secondary or tertiary adrenal insufficiency.
3.ACTH stimulation tests. If the diagnosis of adrenal insufficiency is being considered and the patients have early morning serum cortisol concentration <15 μg/ dL, a short ACTH stimulation test should be performed. A subnormal response confirms the diagnosis of adrenal insufficiency.
4.Corticotropin-releasing hormone test. Differentiation between secondary and tertiary adrenal insufficiency can be done by a corticotropin-releasing hormone test. Patients with secondary adrenal insufficiency show little or no ACTH response, whereas patients with tertiary disease usually have an exaggerated and prolonged ACTH response.
5.Antiadrenal antibodies. Antibodies against 21-hydroxylase (P450c21) are identified in 60–70% of patients with autoimmune adrenal insufficiency. They frequently precede the onset of disease. They are also present in 20% of patients with hypoparathyroidism.
6.Patients with suspected adrenal crisis should be treated with dexamethasone, which does not cross-react in the cortisol assay, and confirmatory tests should be performed within 1–2 days.
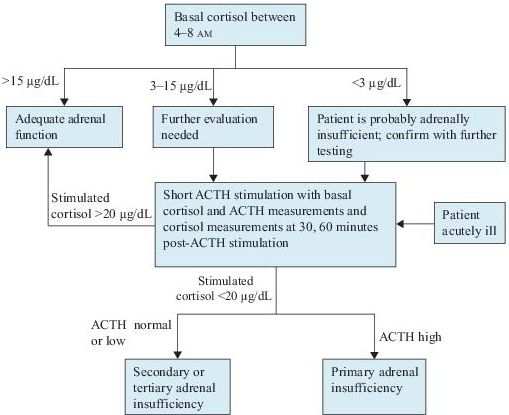
Figure 6–5 Algorithm for the diagnosis of adrenal insufficiency. ACTH, adrenocorticotropic hormone.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


