INTRODUCTION
Operations on the adrenal glands are performed for primary hyperaldosteronism, pheochromocytoma, hypercortisolism (Cushing disease or Cushing syndrome), and adrenocortical carcinoma. These conditions are usually characterized by hypersecretion of one or more of the adrenal hormones. Less commonly, surgery may also be performed for nonfunctioning tumors or metastases.
ANATOMY & SURGICAL PRINCIPLES
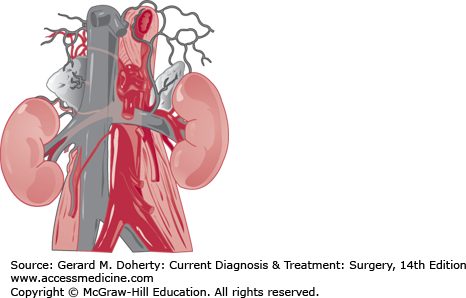
The normal combined weight of the adrenals is 7-12 g. The right gland lies posterior and lateral to the vena cava and superior to the kidney (Figure 33–1). The left gland lies medial to the superior pole of the kidney, just lateral to the aorta and immediately posterior to the superior border of the pancreas. An important surgical feature is the remarkable constancy of the adrenal veins. The right adrenal vein, 2-5 mm long and several millimeters wide, connects the anteromedial aspect of the adrenal gland with the posterolateral aspect of the vena cava. The left adrenal vein is several centimeters long and travels inferiorly from the lower pole of the gland, joining the left renal vein after receiving the inferior phrenic vein. Variant venous drainage (multiple or anomalous veins) occurs in about 5% of glands and are more common for pheochromocytomas and larger tumors. The adrenal arteries are small, multiple, and inconstant. They usually come from the inferior phrenic artery, the aorta, and the upper pole branch of renal artery.
With the exception of rare nonsecreting cancers, indications for adrenal surgery result from hypersecretory states. Diagnosis and treatment begin with confirmation of a hypersecretory state (ie, measurement of excess cortisol, aldosterone, or catecholamines in blood or urine). In order to determine whether the problem originates in the adrenal, levels of the trophic hormone in question (ie, adrenocorticotropic hormone [ACTH] or renin) must be measured. If levels of the trophic hormone are suppressed but hormone secretion is excessive, autonomous secretion is proved. The next step, except in pheochromocytoma, is to determine the degree of autonomy, a process that usually distinguishes hyperplasias (which respond to most but not all controlling mechanisms) from adenomas and adenomas from cancers. In general, cancers are under little if any feedback control. If the primary problem is not in the adrenal, as in Cushing disease, treatment must be directed elsewhere when possible.
The major principles of adrenal surgery are as follows:
Whenever possible, the surgeon must be certain of the diagnosis and the location of the lesion before undertaking the operation.
The patient must be thoroughly prepared so he or she can withstand any metabolic problems caused by the disease or by the operation.
The surgeon and consultants must be able to detect and treat any metabolic crisis that occurs during or after operation.
SURGICAL APPROACHES
Currently, almost all adrenal tumors are identified preoperatively by localization studies such as CT and MRI, so very few operations require general exploration of the abdomen. This permits the use of minimally invasive surgery. Almost all adrenal tumors can be removed laparoscopically. Traditional open adrenalectomy is necessary only when the tumor is especially large (eg, > 8-10 cm, depending on the surgeon’s experience) or for locally invasive adrenocortical cancer where resection of lymph nodes or adjacent organs may be required.
Laparoscopic adrenalectomy can be performed using a transabdominal or retroperitoneal approach. Transabdominal laparoscopic adrenalectomy involves medial rotation of the spleen and pancreas (on the left) or the liver (on the right), using gravity to drop the viscera away from the adrenal. Retroperitoneal adrenalectomy involved the use of high insufflation pressure to maintain the space for dissection and to decrease bleeding during dissection. Although transabdominal approach is more suitable for larger tumors and the retroperitoneal approach better for bilateral adrenalectomy and for patients with abdominal adhesions, surgeon preference usually dictate the choice of surgical approach.
The traditional open surgical approach is used when laparoscopic expertise is not available or when required by the size and invasiveness of the tumor. The advantages of laparoscopic operation are so great that it is strongly preferred.
The open anterior approach (midline, subcostal, or “L” incision) allows wide exposure for large tumors but causes more pain and wound complications, and it requires longer hospitalization. The open posterior approach through the bed of the 11th or 12th rib has been superseded by the laparoscopic approach. Thoracoabdominal incision may be used for large or invasive tumors.
DISEASES OF THE ADRENALS
ESSENTIALS OF DIAGNOSIS
Hypertension with or without hypokalemia.
Elevated aldosterone secretion and suppressed plasma renin activity.
Metabolic alkalosis, relative hypernatremia.
Weakness, polyuria, paresthesias, tetany, cramps due to hypokalemia.
Aldosterone, the most potent mineralocorticoid secreted by the adrenal cortex, regulates the body’s electrolyte composition, fluid volume, and blood pressure. Excess aldosterone increases total body sodium, decreases potassium levels, increases extracellular fluid volume (without edema), and increases blood pressure. Under normal conditions, aldosterone secretion is regulated by the renin-angiotensin system in a feedback fashion and is also stimulated transiently by ACTH.
In primary aldosteronism, aldosterone levels are elevated and renin levels are suppressed. In secondary hyperaldosteronism, increased aldosterone is due to increased renin secretion. Examples of secondary hyperaldosteronism include renovascular disease, renin-secreting tumors, and cirrhosis with low intravascular volume or diuretic use. Among the subtypes of primary aldosteronism, aldosterone-producing adenoma (APA, aldosteronoma) and idiopathic hyperaldosteronism (IHA) with adrenal hyperplasia are the most common types. Unilateral primary adrenal hyperplasia, aldosterone-producing adrenocortical carcinoma, and familial hyperaldosteronism (FH) (eg, FH type I-glucocorticoid-remediable hyperaldosteronism and FH type II) are rare. Surgery is beneficial only in patients with APAs and in patients with unilateral primary adrenal hyperplasia.
Primary aldosteronism in its classic form is characterized by hypertension, hypokalemia, increased aldosterone secretion, and suppressed plasma renin activity (PRA). However, hypokalemia is not required to make the diagnosis; recent studies have shown that many patients have a normal potassium level. Primary aldosteronism was once thought to be present in about 1% of patients with hypertension, but its prevalence has increased to 5%-13% based on various studies when plasma aldosterone concentration (PAC)-to-PRA was used to screen for hyperaldosteronism in patients with hypertension who were not hypokalemic. Although rare, normotensive primary aldosteronism has been described.
Aldosteronomas are usually solitary and small (0.5-2 cm). They have a characteristic golden-yellow color when sectioned. Tumor cells typically have heterogeneous cytomorphology, resembling those of all three zones of the adrenal cortex, including hybrid cells having cytologic features of the zona glomerulosa and zona fasciculata. Hyperplasia is also often seen in glands harboring adenomas.
Aldosterone facilitates the exchange of sodium for potassium and hydrogen ions in the distal nephron. Therefore, when aldosterone secretion is chronically increased, serum potassium and hydrogen ion concentrations fall (hypokalemia and alkalosis), total body sodium rises, and hypertension results.
Symptoms, if present, are usually those of hypokalemia and depend on the severity of potassium depletion. Patients complain of a sense of malaise, muscle weakness, polyuria, polydipsia, cramps, and paresthesias. Tetany and hypokalemic paralysis occur rarely. Headaches are common. Hypertension is usually moderate to severe and may be refractory to medical therapy, but advanced retinopathy is rare. Although extracellular fluid volume is increased, edema is not seen unless renal failure occurs.
Primary aldosteronism should be suspected in patients with hypertension and hypokalemia—either spontaneous or following the administration of diuretics—and in patients with refractory hypertension. The diagnostic evaluation should start with screening tests. A simple ambulatory test determines the ratio of PAC, in nanograms per deciliter, to PRA, in nanograms per milliliter per hour, performed in the morning in a seated ambulant patient. A ratio greater than 20 with a PAC greater than 15 ng/dL suggests primary aldosteronism and warrants confirmatory biochemical studies. Hypertensive individuals without primary aldosteronism usually have ratios of less than 20. If the patient is taking an aldosterone receptor antagonist spironolactone or eplerenone, the data are uninterpretable, and estrogens increase PACs by increasing angiotensinogen. These agents should be discontinued for 6 weeks before the workup.
Many medications affect PAC and PRA. For example, PAC is decreased by angiotensin-converting enzyme inhibitors (ACE inhibitors), angiotensin receptor blockers (ARBs), central alpha-2 agonists such as clonidine, dihydropyridine (DHP) calcium channel antagonists such as amlodipine, and beta-blockers. PRA is increased by ACE inhibitors, ARBs, and DHP-calcium channel antagonists. These medications should be discontinued for 2 weeks if necessary. Peripheral α-adrenergic blockers such as doxazosin, terazosin, prazosin, hydralazine, and non-DHP calcium channel blocker such as verapamil are the preferred antihypertensive agents during evaluation. In many patients, it is unwise to withdraw antihypertensive medications, and one must be content with imperfect data.
If the screening test is positive, failure to suppress aldosterone secretion with sodium loading will confirm the diagnosis of primary aldosteronism in most patients. Normally aldosterone can be suppressed by oral salt loading or intravenous sodium chloride infusion. The patient should consume a high-sodium diet (5000 mg of sodium for 3 days) or be supplemented with NaCl tablets (2-3 g with each meal) if necessary. A 24-hour urine sample is collected for aldosterone and sodium on the third day. Serum potassium should be monitored because the high-salt diet increases kaliuresis, and potassium chloride should be supplemented to avoid hypokalemia, which interferes with the test results by decreasing aldosterone secretion and may cause cardiac arrhythmias. Urinary aldosterone excretion higher than 12-14 μg/24 h distinguishes most patients with primary aldosteronism from those with essential hypertension on a high-salt diet, as confirmed by urinary sodium excretion exceeding 200 mEq/24 h. Alternatively, a PAC higher than 10 ng/mL after an infusion of 2 L of normal saline over 4 hours is also consistent with primary aldosteronism.
Once the diagnosis is established, the surgically correctable forms—APA (aldosteronoma), constituting approximately 35% of primary aldosteronism, and the rare unilateral primary adrenal hyperplasia—should be distinguished from IHA, comprising approximately 65% of primary aldosteronism, due to bilateral adrenal hyperplasia, for which medical therapy is the best management. Aldosteronoma and IHA are the most common subtypes. Compared with those with IHA, patients with aldosteronoma have more severe hypertension, more severe hypokalemia, higher aldosterone secretion (> 20 ng/dL), higher 18-hydroxycorticosterone concentrations (> 100 ng/dL), and are younger. Interestingly more than one third of aldosteronomas have a somatic KCNJ5 gene mutation.
Glucocorticoid-remediable hyperaldosteronism (FH type I) is inherited in an autosomal dominant fashion. The genetic defect results in a chimeric gene. The mutated gene juxtaposes the promoter for expression of the 11-hydroxylase gene, which is ACTH-responsive, with the coding sequence of the aldosterone synthase gene. This leads to aldosterone production under ACTH stimulation in the zona fasciculata. Glucocorticoid therapy reverses this type of hyperaldosteronism. These patients have a family history of onset of hypertension at an early age. The diagnosis can be established by genetic testing. Measurement of 24-hour urine 18-hydroxycortisol and 18-oxocortisol levels is less reliable. The molecular basis for FH type II is not clear although it is also inherited in an autosomal dominant fashion.
Aldosterone-secreting adrenocortical carcinoma is rare and should be suspected if the tumor is larger than 4 cm, especially if it also secrets cortisol and intermediate metabolites.
An APA can frequently be demonstrated by high-resolution CT or MRI scanning. Small aldosteronomas can be missed, and in such cases, a patient with a small aldosteronoma not seen on CT may be misdiagnosed as having adrenal hyperplasia. Aldosteronomas that coexist with nonfunctional adenomas can be mislabeled as adrenal hyperplasia because of multinodularity or bilateral masses on CT. Small abnormalities on CT scans may represent hyperplasia rather than true aldosteronomas. Therefore, unless an unequivocal unilateral tumor, preferably larger than 1 cm, is present on the CT scan and the contralateral gland is normal, the diagnosis and localization of aldosteronoma is not certain. When in doubt, adrenal vein sampling should be done. Blood is sampled from the adrenal veins and the inferior vena cava for aldosterone and cortisol levels at baseline and after ACTH infusion. Proper catheter placement is confirmed by finding high cortisol levels in adrenal venous blood compared with the inferior vena cava. Corrected aldosterone levels are calculated from the ratio of aldosterone to cortisol in each venous sample. A lateralization ratio of the corrected aldosterone level higher than 4 indicates unilateral aldosterone secretion, thereby confirming a diagnosis of aldosteronoma in most patients. Adrenal vein sampling is invasive and requires considerable skill and experience. The success rate for cannulating both adrenal veins is about 90% (60%-95% depending on experience of radiologists). Complication of venous sampling, such as adrenal hemorrhage can occur in 1% of patients. Some recommend adrenal vein sampling to be done routinely regardless of CT findings. Whereas others advocate using it selectively when CT scan findings are uncertain (no tumor, bilateral tumors, or small tumor < 1 cm in diameter).
Uncontrolled hypertension can lead to renal failure, stroke, and myocardial infarction. Severe hypokalemia can cause weakness, paralysis, and arrhythmia, especially in patients taking digitalis.
The goal of therapy is to prevent the complications of hypertension and hypokalemia. Unilateral adrenalectomy is recommended for patients with aldosteronoma and medical therapy for those with IHA or those with aldosteronoma who are poor candidates for surgery.
Blood pressure and hypokalemia should be controlled before surgery. Spironolactone, a competitive aldosterone antagonist, has been the drug of choice. It blocks the mineralocorticoid receptor, promotes potassium retention, restores normal potassium concentrations, and reduces the extracellular fluid volume, thereby controlling blood pressure. Furthermore, it reactivates the suppressed renin-angiotensin-aldosterone system in the contralateral adrenal gland, reducing the risk of postoperative hypoaldosteronism. The medication should be continued to the day of operation. For additional information and medical regiments for preoperative preparation, see Medical Treatment section.
Because aldosteronomas are almost always small and benign, laparoscopic adrenalectomy is the procedure of choice. It can be performed safely and with equally good results by several approaches. The lateral transabdominal approach uses gravity to help medially rotate the viscera (liver on the right and spleen and pancreas on the left) and exposes the adrenal gland. It is the most versatile approach since the anatomy is familiar to most abdominal surgeons. The posterior retroperitoneal approach is best in patients with prior upper abdominal operations, but the working space is more limited. Although some surgeons perform a subtotal resection for aldosteronoma, most excise the whole adrenal gland with the tumor. The surrounding adrenal tissue frequently appears hyperplastic. Small aldosteronomas may not be visible intraoperatively. Bilateral adrenalectomy is not indicated, since hypocortisolism will require steroid-replacement treatment. Patients with IHA should be treated medically.
Occasional patients may develop transient aldosterone deficiency because of suppression of the contralateral adrenal gland by the hyperfunctioning adenoma. This is rare in patients treated with spironolactone preoperatively. Symptoms include postural hypotension and hyperkalemia. Adequate sodium intake is usually sufficient for treatment; rarely, short-term fludrocortisone replacement (0.1 mg/d orally) is required.
The goal is to control hypertension and hypokalemia. Spironolactone, a competitive aldosterone antagonist, has been the drug of choice. Initial dosages of 200-400 mg/d may be required to control hypokalemia and hypertension. Once blood pressure is normalized and hypokalemia is corrected, the dose can be tapered and maintained at about 100-150 mg/d. Spironolactone may have antiandrogenic side effects, such as impotence, gynecomastia, menstrual irregularity, and gastrointestinal disturbances. These potential side effects may make this medication less desirable for some patients. Unlike spironolactone, which also blocks androgen and progesterone receptors, eplerenone is a selective mineralocorticoid receptor antagonist and has fewer endocrine side effects. It has been approved for treatment of hypertension and for heart failure after myocardial infarction. Eplerenone may become the treatment of choice for primary aldosteronism because of its decreased side effects if it is proven as efficacious as spironolactone, in spite of its increased cost. Amiloride, 20-40 ng/d, a potassium-sparing diuretic, may be used alternatively or as a supplement to spironolactone. Other medications, such as ACE inhibitors, calcium-channel blockers, and diuretics, may be required to control hypertension.
Primary aldosteronism usually follows a prolonged and subtly changing course. Untreated hypertension may cause stroke, myocardial infarction, or renal failure.
Removal of an aldosteronoma normalizes potassium levels in nearly 100% of the patients, but hypertension is cured in about 50% of the patients. Persistent hypertension after operation is more common in overweigh men who preoperatively required more than three antihypertensive medications for more than 6 years, but residual hypertension is usually easier to control than before the operation. Essential hypertension and atherosclerosis due to chronic hypertension are contributing factors. Although patients with IHA should be treated medically, adrenalectomy is indicated for those with aldosteronoma because side effects of the medications and compliance make long-term medical treatment undesirable. The low morbidity, short hospitalization, and high success rate of laparoscopic adrenalectomy have made surgery preferable to long-term medical therapy.
PHEOCHROMOCYTOMA
ESSENTIALS OF DIAGNOSIS
Hypertension, frequently sustained, with or without paroxysms.
Episodic headache, excessive sweating, palpitation, and visual blurring.
Postural tachycardia and hypotension.
Elevated urinary catecholamines or their metabolites, hypermetabolism, hyperglycemia.
Pheochromocytomas are tumors of the adrenal medulla and related chromaffin tissues elsewhere in the body (paragangliomas) that secrete epinephrine or norepinephrine, resulting in sustained or episodic hypertension and other symptoms of catecholamine excess.
Pheochromocytoma is found in less than 0.1% of patients with hypertension and accounts for about 5% of adrenal tumors incidentally discovered by CT scanning. Most pheochromocytomas occur sporadically without other diseases, but about one third are associated with various familial syndromes such as MEN2, NF1, VHL, and familial paraganglioma syndromes. Patients with multiple endocrine neoplasia (MEN)2A may have medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism. Those with MEN2B have medullary thyroid carcinoma, pheochromocytoma, mucosal neuromas, marfanoid habitus, and ganglioneuromatosis. Patients with neurofibromatosis type I have café au lait spots, neurofibromatosis, and pheochromocytoma. Patients with von Hippel–Lindau disease may have retinal hemangioma, hemangioblastoma of the central nervous system, renal cysts and carcinoma, pheochromocytoma, pancreatic cysts or neuroendocrine tumors, and epididymal cystadenoma. Familial paraganglioma syndromes are caused by mutations of the succinate dehydrogenase genes SDHx (SDHB, SDHD, SDHC, SDHA, and SDHAF2). All are associated with extra-adrenal paragangliomas. SDHB is particularly associated with malignant pheochromocytomas. These syndromes should be considered especially in young patients and in patients with multifocal, extra-adrenal, malignant, or recurrent tumors. Other genetic mutations associated with pheochromocytomas are TMEM127 and MAX. Because of the higher than previously expected prevalence of hereditary pheochromocytoma, all patients with pheochromocytoma should be considered for genetic counseling and testing. The specific genes tests should depend on clinical and biochemical findings and family history. Family members of patients who have been diagnosed with these syndromes also need screening to determine whether they are gene carriers and are at risk for developing the various tumors, including pheochromocytoma.
On pathologic examination, pheochromocytoma appears reddish-gray and frequently has areas of necrosis, hemorrhage, and sometimes cysts. The usual size is about 100 g, or 5 cm in diameter, but they can be as small as 2-3 cm or as large as 12-16 cm. Cells are pleomorphic, showing prominent nucleoli and frequent mitoses. Cytologic findings cannot be used to determine whether a pheochromocytoma is malignant or benign. The veins and capsules may also be invaded even in clinically benign tumors. Malignancy can only be diagnosed in the presence of metastases in nonchromoffin cell bearing tissues or invasion into surrounding tissues.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


