ANATOMY
The spleen is a dark purplish, highly vascular, coffee bean-shaped organ of mesodermal origin situated in the left upper quadrant of the abdomen at the level of the eighth to eleventh ribs between the fundus of the stomach, the diaphragm, the splenic flexure of the colon, and the left kidney (Figure 27–1). The adult spleen weighs 100-150 g, measures about 12 × 7 × 4 cm, and usually cannot be palpated. It is attached to adjacent viscera, the abdominal wall, and the diaphragm by peritoneal folds or “ligaments.” The gastrosplenic ligament carries the short gastric vessels. The other ligaments are avascular except in patients with portal hypertension or myelofibrosis.
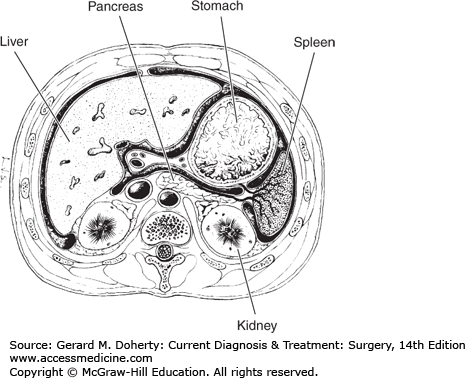
The splenic capsule consists of peritoneum overlying a 1- to 2-mm fibroelastic layer that contains a few smooth muscle cells. The fibroelastic layer sends into the pulp numerous fibrous bands (trabeculae) that form the framework of the spleen. Corrosion cast studies demonstrate that the spleen consists of specific segments based on arterial supply numbering between two and six separated by an avascular plane.
The splenic artery enters the hilum of the spleen, branches into the trabecular arteries, and then branches into the central arteries that course through the surrounding white pulp and send radial branches to the peripheral marginal zone and the more distant red pulp. The white pulp consists of lymphatic tissue including T cells adjacent to the central artery (periarteriolar lymphoid sheets [PALS]), with a surrounding area containing lymphoid follicles rich in B cells interspersed with dendritic and reticular cells important in antigen presentation. The vascular spaces of the marginal zone between the red and white pulp channel blood into the splenic Billroth cords and out to the associated sinuses. The red pulp vascular structures have a noncontiguous basement membrane that filters cells such as senescent erythrocytes into the macrophage-lined sinuses.
Accessory spleens (spleniculi) are seen in 10%-15% of the normal population and are located primarily in the gastrosplenic, gastrocolic, and lienorenal ligaments, but they can also be found throughout the peritoneal cavity in the omentum, bowel mesentery, and pelvis. Accessory spleens probably result from a failure of infusion of splenic embryologic tissues. Ordinarily of no significance, they may play a role in recurrence of certain hematologic disorders for which splenectomy is performed. Removal of accessory spleens may lead to remission of disease in these patients. Accessory spleens are more difficult to identify with laparoscopic procedures, but the use of a hand port has allowed identification and resection of accessory spleens with a minimally invasive approach. Patients who fail to respond to initial splenectomy should undergo scanning with technetium 99m-labeled red cells or indium 111-labeled platelets to identify potential sites of missed accessory spleens and can be identified intraoperatively with a hand-held gamma counter.
Ectopic spleen (wandering spleen) is an unusual condition in which a long splenic pedicle allows the spleen to move within the peritoneum. It often resides in the lower abdomen or pelvis, where even a normal-sized spleen can be felt as a mass. The condition is 13 times more common in women than in men. Diagnostic radionuclide scan can diagnose the mass as a spleen. Acute torsion of the pedicle occurs occasionally, necessitating emergency splenectomy, and elective removal of wandering spleens in the pelvis is recommended.
PHYSIOLOGY
The spleen has a dual function as a secondary lymphoid organ important in host immunity, and as a large filter for blood removing senescent erythrocytes and recycling iron. The anatomy of the spleen provides an ideal environment for these two functions with the immune activity in the white pulp and the hematologic function in the red pulp.
The spleen receives 5% of the total cardiac output, or approximately 150-300 mL/min, such that each red cell averages 1000 passes through the spleen each day. Normal blood cells pass rapidly through the spleen, while abnormal and senescent cells are slowed and entrapped. As they travel through the hypoxic, acidotic, glucose-deprived splenic cords and sinuses of the red pulp, senescent erythrocytes pass into the vascular spaces and are phagocytosed by macrophages in a process called “culling.” Part of the membrane of erythrocytes can be removed through the gaps between endothelial cells lining the vascular spaces in a similar process called pitting. In the presence of splenomegaly and other disease states, the flow patterns of the spleen become more circuitous as the red pulp volume expands, so that even normal cells may be trapped.
The spleen is considered to be a secondary organ of the immune system and represents the largest single collection of lymphoid tissue in the body. The white pulp of the spleen contains the various cellular components needed to generate an immune response, with structural and functional relationships similar to those of lymph nodes. Lymphocytes and circulating antigen-presenting cells enter the white pulp via the marginal zone capillaries and traverse the T cell-rich PALS before passing through bridging channels into the red pulp. Primary follicles or germinal centers with secondary follicles at the periphery of the white pulp are sites of B cell expansion and immunoglobulin production. Blood passing through the spleen is exposed to all the key cellular components necessary for both humoral and cellular immune responses. Tissue macrophages in the spleen are key components of generating an immune response particularly in encapsulated organs. Moreover, the concentration of macrophages in the red pulp vascular spaces facilitates opsonization of particles coated with IgG and plays an important role in the filtration and removal of senescent erythrocytes, the autoimmune hematologic diseases as well as explaining the increased risk of sepsis that follows splenectomy in children under 2 years of age. Even in adults, splenectomy leads to a slight but definite reduction in immune function.
Normally, about 30% of the total platelet pool is sequestered in the spleen. Splenomegaly typically involves expansion of the red pulp, which increases this sequestration to between 80% and 95% of the platelet cell mass. Storage of erythrocytes and granulocytes in the spleen is limited in humans, but newly formed reticulocytes released from the bone marrow concentrate in the spleen to undergo a maturational process.
OPERATIVE INDICATIONS FOR SPLENECTOMY
To better describe and understand operative indications in surgery of the spleen, one could categorize the indications for splenectomy or procedures of the spleen into eight general areas:
Hypersplenism is characterized by diffuse enlargement of the spleen by neoplastic disorders, hematopoietic disorders of the bone marrow, and metabolic or storage disorders. These various disease processes result in diffuse enlargement of the spleen and amplify the normal function of elimination of circulating blood cells resulting in general pancytopenia. Erythrocytes and platelets are most commonly affected. Hypersplenism also may cause symptoms of early satiety due to the splenic size.
Autoimmune/erythrocyte disorders Specific cytopenias are related either to antibodies targeting platelets, erythrocytes, or neutrophils. A second category of diseases relates to intrinsic structural changes within the erythrocyte that lead to a shortened red blood cell half-life with accelerated splenic clearance. There is nothing intrinsically wrong with the spleen, and splenic size is typically normal.
Trauma or injury to the spleen.
Vascular diseases Splenic vein thrombosis and splenic artery aneurysm may require splenectomy for treatment.
Cysts, abscesses, and primary splenic tumors are mass lesions of the spleen. This would include treatment of simple cysts, echinococcal cysts, splenic abscess, and various benign neoplasms including hamartomas, hemangiomas, lymphangiomas, and rare malignant lesions.
Diagnostic procedures This category of splenectomy occurs when the spleen is removed primarily to make a clinical diagnosis when none is available. A subcategory of this would be staging laparotomy for Hodgkin disease, which has all but been eliminated based on alternative imaging techniques and current treatment regimens.
Iatrogenic splenectomy Splenectomy that is performed due to an incidental injury to the spleen during surgery within the general abdominal cavity or specifically, the left upper quadrant, can be categorized as iatrogenic splenectomy. This category is likely underreported and may be considered a subcategory of trauma.
Incidental splenectomy The spleen may be removed as part of a standard operation to remove the distal pancreas most commonly, and also for gastric cancers, left-sided renal cell carcinomas, adrenal cancers, and retroperitoneal sarcomas in the left upper quadrant. The spleen is removed in these instances due to direct tumor extension, vascular involvement, or the need for excision of splenic hilum lymph nodes.
With the increase in splenic preservation for trauma, many institutional series list medical conditions as the most indications for splenectomy. Most recent series report 40%-50% for hematologic conditions, 35%-40% for trauma, and 20%-30% for neoplastic disease. Within the category, idiopathic thrombocytopenia purpura has the highest incidence of splenectomy. Each of these categories of disease will be discussed including the etiology and pathophysiology of the disorder, the specific indications for splenectomy, alternative treatments, and the results of splenectomy.
In the past, the term hypersplenism or increased splenic function has been used to denote the syndrome characterized by splenic enlargement, deficiency of one or more blood cell lines, normal or hyperplastic cellularity of deficient cell lines in the marrow, and increased turnover of affected cells. Increased understanding of the pathophysiology of specific disorders has shown that hypersplenism is not synonymous with splenomegaly. Some disorders in which there is spleen-dependent destruction of blood elements do not manifest all features of hypersplenism. For example, splenomegaly is rarely a feature of immune thrombocytopenic purpura, and splenectomy is not always curative. Conversely, other conditions that enlarge the spleen may not result in destruction or sequestration of blood elements with resultant cytopenias. In disorders with known pathogenesis, the trend has been to classify them as separate disease entities rather than as hypersplenic conditions.
The defects in hypersplenism are exaggerations of normal splenic functions primarily associated with the red pulp. The principal cause of cytopenias in hypersplenism is increased sequestration and destruction of blood cells in the spleen, which is hypertrophied or increased in volume in a variety of diseases. Etiologic factors include: (1) neoplastic infiltration, (2) disease of the bone marrow in which the spleen becomes a site of extramedullary hematopoiesis, or (3) metabolic/genetic disorders such as Gaucher disease. The hyperplastic spleen is not selective in its hyperfunction in most of these disorders. The splenomegaly can lead to an increased turnover in erythrocytes and platelets, with a lesser effect on leukocytes. For example, about 60% of patients with cirrhosis develop splenomegaly and 15% develop hypersplenism. The hypersplenism of cirrhosis is seldom of clinical significance; the anemia and thrombocytopenia are usually mild and rarely are indications for splenectomy.
The clinical findings depend largely on the underlying disorder or are secondary to the depletion of circulating blood elements caused by the hypersplenism (Table 27–1). Manifestations of hypersplenism usually develop gradually, and the diagnosis often follows a routine physical or laboratory examination. Some patients experience left upper quadrant fullness, discomfort (can be severe), or early satiety. Others have hematemesis due to gastroesophageal varices.
| Congestive splenomegaly (cirrhosis, portal or splenic vein obstruction) |
| Neoplasm (leukemia, metastatic carcinoma) |
| Inflammatory disease (sarcoid, lupus erythematosus, Felty syndrome) |
| Acute infections with splenomegaly |
| Chronic infection (tuberculosis, brucellosis, malaria) |
| Storage diseases (Gaucher disease, Letterer–Siwe disease, amyloidosis) |
| Chronic hemolytic diseases (spherocytosis, thalassemia, glucose-6-phosphate dehydrogenase deficiency, ellipto-cytosis) |
| Myeloproliferative disorders (myelofibrosis with myeloid metaplasia) |
Purpura, bruising, and diffuse mucous membrane bleeding are unusual symptoms despite the presence of thrombocytopenia. Anemia may produce significant fatigue that may be the chief complaint in this patient population. Recurrent infections may be seen in patients with severe leukopenia.
Patients with primary hypersplenism usually exhibit pancytopenia of moderate degree and generalized marrow hyperplasia. Anemia is most prominent, reflecting the destruction of erythrocytes in the hypertrophied red pulp of the spleen. Thrombocytopenia occurs due to sequestration of platelets but also possibly due to increased turnover. In most cases more immature cell types such as reticulocytes are present, reflecting the overactivity of the bone marrow to compensate for the pancytopenias. One exception is myeloid metaplasia, in which dysfunction of the bone marrow is the primary defect.
Before it becomes palpable, an enlarged spleen may cause dullness to percussion above the left costal margin. Splenomegaly is manifested on supine x-rays of the abdomen by medial displacement of the stomach and downward displacement of the transverse colon and splenic flexure. CT scan is useful for differentiating the spleen from other abdominal masses and for demonstrating splenic enlargement or intrasplenic lesions. Some of the largest massive spleens (spleen weight > 1500 g) occur in these types of disease. Finding the edge of the spleen below the iliac crest and crossing the abdominal midline are frequently seen.
Leukemia and lymphoma are diagnosed by marrow aspiration, lymph node biopsy, and examination of the peripheral blood (white count and differential). In hereditary spherocytosis there are spherocytes, osmotic fragility is increased, and platelets and white cells are normal. The hemoglobinopathies with splenomegaly are differentiated on the basis of hemoglobin electrophoresis or the demonstration of an unstable hemoglobin level. Thalassemia major becomes apparent in early childhood, and the blood smear morphology is characteristic. In myelofibrosis, the bone marrow shows proliferation of fibroblasts and replacement of normal elements. In idiopathic thrombocytopenic purpura (ITP), the spleen is normal or only slightly enlarged. In aplastic anemia, the spleen is not enlarged and the marrow is fatty.
The course, response to treatment, and prognosis of the hypersplenic syndromes differ widely depending on the underlying disease and its response to treatment and will be discussed for each particular disorder below. The indications for splenectomy are given in Table 27–2.
| Splenectomy Always Indicated |
| Primary splenic tumor (rare) |
| Hereditary spherocytosis (congenital hemolytic anemia) |
| Splenectomy Usually Indicated |
| Primary hypersplenism |
| Chronic immune thrombocytopenic purpura |
| Splenic vein thrombosis causing gastric varices |
| Splenic abscess (rare) |
| Splenectomy Sometimes Indicated |
| Splenic injury |
| Autoimune hemolytic disease |
| Elliptocytosis with hemolysis |
| Nonspherocytic congenital hemolytic anemias |
| Hodgkin disease (for staging) |
| Thrombotic thrombocytopenic purpura |
| Idiopathic myelofibrosis |
| Splenic artery aneurysm |
| Wiscott–Aldrich syndrome |
| Gaucher disease |
| Mastocytosis-aggressive disease |
| Splenectomy Rarely Indicated |
| Chronic leukemia |
| Splenic lymphoma |
| Macroglobulinemia |
| Thalassemia major |
| Sickle cell anemia |
| Congestive splenomegaly and hypersplenism due to portal hypertension |
| Felty syndrome |
| Hairy cell leukemia |
| Chédiak–Higashi syndrome |
| Sarcoidosis |
| Splenectomy Not Indicated |
| Asymptomatic hypersplenism |
| Splenomegaly with infection |
| Splenomegaly associated with elevated IgM |
| Hereditary hemolytic anemia of moderate degree |
| Acute leukemia |
| Agranulocytosis |
Splenectomy may decrease transfusion requirements, decrease the incidence and number of infections, prevent hemorrhage, and reduce pain. The course of congestive splenomegaly due to portal hypertension depends upon the degree of venous obstruction and liver damage. The hypersplenism is rarely a major problem and is almost always overshadowed by variceal bleeding or liver dysfunction.
Neoplastic diseases in which splenectomy may play a role in the management of hypersplenism include chronic lymphocytic leukemia (CLL), hairy cell leukemia, and non-Hodgkin lymphoma. Lymphoma is discussed in detail in Chapter 44. Related neoplastic disorders of idiopathic myelofibrosis and mastocytosis are also discussed as precursors or variants of neoplastic diseases in which splenectomy are occasionally indicated.
CLL is a low-grade neoplasm of B cell lineage characterized by accumulations of populations of lymphocytes that are mature morphologically but functionally incompetent. In the United States, CLL occurs as 25%-30% of all leukemias with mean age at diagnosis of 72. The clinical manifestations and natural history are variable, but initially the disease tends to be indolent. In more advanced stages, splenomegaly, which is frequently massive, is a common characteristic of CLL. Most symptoms related to the spleen are from thrombocytopenia and anemia due to secondary hypersplenism (80%-90% of splenic symptoms). Ten to 20% of patients may have symptoms primarily related to pressure from the size of the enlarged spleen.
Other causes of cytopenia in CLL relate to decreased cellular production from the bone marrow. Bone marrow failure can be due to replacement with leukemic cells or to depletion of the bone marrow as a toxic effect of prior antitumor chemotherapy.
Splenectomy in patients with CLL corrects thrombocytopenia in 70%-85% of cases, neutropenia in 60%-70%, and anemia in 50%-60% of cases. The median duration of benefit for both platelets and red cell populations is well over 1 year. Patients with smaller spleens preoperatively, lower preoperative platelet counts, and extensive prior chemotherapy are less likely to respond to splenectomy. However, a positive bone marrow aspirate for leukemic cells is not a contraindication to splenectomy in CLL. Patients who do not have a good performance status should not undergo splenectomy, since patients in terminal stages have unacceptable operative morbidity.
Hairy cell leukemia is a low-grade lymphoproliferative disorder with characteristic “hairy cells”—ie, B lymphocytes with irregular cytoplasmic protrusions positive for tartrate reaction acid phosphatase—which infiltrate the bone marrow and spleen. Patients are typically male, and onset of the disease is in the fifth or sixth decade of life. Symptoms relate to pancytopenia, with anemia requiring transfusions; and to neutropenia, characterized by increased susceptibility to infections and increased bleeding tendencies. Some patients may have symptoms from splenomegaly, which is present in 80% of patients at the time of diagnosis of hairy cell leukemia. The cytopenias are due to a combination of bone marrow replacement and secondary hypersplenism.
The standard therapy for hairy cell leukemia between 1960 and 1995 was splenectomy, but recent advances in pharmacotherapy have superseded this surgical approach. First-line therapy is now treatment with purine nucleoside analogues primarily cladribine, with a complete response rate of 80%-90%. It has never been shown that splenectomy offers survival benefit in this indolent disease, and the operation should be reserved for palliation of splenomegaly in patients who have failed treatment with cladribine and second line agent rituxamib and alpha interferon.
Myelodysplastic syndromes are a heterogeneous group of clinical hematopoietic stem cell disorders manifested by pancytopenias, and dysplasia of the bone marrow. Pathologic changes include extensive bone marrow fibrosis, extramedullary hematopoiesis in the spleen and liver, and a leukoerythroblastic blood reaction that may evolve into acute myeloid leukemia over time.
The bone marrow is usually almost completely replaced by fibrous tissue, although in some cases it is hyperplastic and fibrosis is minimal. Extramedullary hematopoiesis develops mainly in the spleen, liver, and long bones. Symptoms are attributable to anemia (weakness, fatigue, dyspnea) and to splenomegaly (abdominal fullness and pain, which may be severe). Pain over the spleen from splenic infarcts is common. Spontaneous bleeding, fatigue, secondary infection, bone pain, and a hypermetabolic state are frequent. Portal hypertension develops in some cases as a result of fibrosis of the liver, greatly increased splenic blood flow, or both.
Hepatomegaly is present in 75% of cases and splenomegaly with a firm and irregular spleen in all cases. Striking changes in the peripheral blood are referable to the combination of extramedullary hematopoiesis and hypersplenism. Patients are uniformly anemic, and red cells vary greatly in size and shape, many of them distorted and fragmented. The white count is usually high (20,000-50,000/mL). The platelet count may be elevated, but values less than 100,000/mL are seen in 30% of cases due to secondary hypersplenism. Bone marrow aspirates frequently result in a dry tap because marrow is replaced with fibrosis. It was once incorrectly thought that the spleen performed a crucial function of extramedullary hematopoiesis in this disease and that splenectomy could be lethal. In fact, many patients with myeloid metaplasia feel better if the massive spleen is removed, and their hypersplenism is often corrected.
About 30% of patients are asymptomatic at the time of initial diagnosis and require no therapy. When cytopenias and splenomegaly produce symptoms, treatment is primarily supportive using transfusions, androgenic steroids, antimetabolites, and hematopoietic growth factors are indicated. Newer therapies include treatment with immunomodulatory drugs such as thalidomide or antibodies to VEGF and TNF. A subset of patients with myeloid metaplasia has a component of autoimmune hemolytic anemia, and in this group of patients immunosuppressive therapy may be beneficial. Splenectomy is indicated in the following situations: (1) major hemolysis unresponsive to medical management, (2) severe symptoms of massive splenomegaly with mass effect of the spleen, (3) life-threatening thrombocytopenia, and (4) portal hypertension with variceal hemorrhage. This is one of the rare occasions when portal hypertension may be cured by splenectomy.
Splenectomy in myeloid metaplasia is associated with a 7%-10% death rate and frequent complications often related to postsplenectomy hepatic morbidity. Splenectomy best relieves symptoms of splenomegaly and portal hypertension, but only about 75% of patients get relief from anemia and thrombocytopenia. Younger patients with normal platelet counts and symptoms are the best candidates for splenectomy in idiopathic myelofibrosis.
Systemic mast cell disease, or mastocytosis, is a rare condition characterized by mast cell infiltration of a number of tissues, including the spleen. There are two types: indolent and aggressive. In indolent systemic mass cell disease, there is no need for consideration of splenectomy. The aggressive type is associated with hematologic diseases with characteristics of lymphoma. Splenomegaly may occur, with the predominant symptoms resulting from thrombocytopenia due to hypersplenism. In this subgroup of patients with aggressive disease, splenectomy improves platelet counts and is associated with longer median survival time than for patients with aggressive disease who do not undergo splenectomy, although systemic therapy including alpha interferon has been shown to be effective.
Metabolic disorders amenable to splenectomy are rare inherited diseases that include as a component splenic enlargement due to the pathologic deposition of material within the spleen. In Gaucher disease, excess sphingolipid is deposited in the spleen. In sarcoidosis, the spleen becomes involved with noncaseating granulomas as can be seen in lymph nodes. Inherited disorders also include disease in which there is a specific immunologic target with associated destruction in the spleen.
Gaucher disease is an autosomal recessive disorder characterized by a deficiency in beta-glucosidase, a lysosomal enzyme that degrades the sphingolipid glucocerebroside. There is an increased incidence of this disorder in Ashkenazi Jews. Three types of this disease exist, and the one amenable to splenectomy is type I, or the adult type. Pathologically, Gaucher disease results in lipid accumulation within the white pulp of the spleen, the liver, or the bone marrow. Predominant symptoms relate to massive splenomegaly from either the direct effects of the size of the spleen or secondary to cytopenias from hypersplenism.
Treatment by total splenectomy alleviates the symptoms but results in accelerated hepatic and bone disease as well as a significant increased risk of postsplenectomy infections. Treatment with partial or subtotal splenectomy has been studied over the past 10 years for both adults and children with Gaucher disease. Removing most of the spleen corrects the symptoms of splenomegaly, but leaving a splenic remnant provides a site for further deposition of lipid that protects the liver and bone. The major problem with partial splenectomy is the eventual recurrence and enlargement of the splenic remnant accompanied by recurrent symptoms. As with hereditary spherocytosis, there is an increase incidence of pigmented gallstones occurring in up to two-thirds of female patients and one-third of male patients. The goal of subtotal splenectomy in Gaucher disease is to leave a small fragment approximately the size of the fist of the patient. Replacement therapy with recombinant glucocerebrosidase enzyme has recently become available, but the cost of chronic treatment is prohibitive.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


