Absolute bioavailability is the amount of drug that is systemically absorbed and pharmacologically active when administered by any route other than IV administration. The following equation allows you to determine absolute bioavailability.
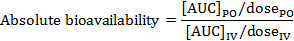
Bioequivalent drugs
The FDA requires that all generic drugs that would like to come to market have both in vitro and in vivo tests performed to prove that they are equivalent to the bioavailability of the original brand name drug. For a generic drug to be FDA approved as bioequivalent, it does not have to be identical in all pharmacokinetic parameters to the original brand drug. It does, however, have to be identical in all parameters that are considered important for proper action of the drug in the body.
The FDA gives all generic drugs that enter the market either an A rating or a B rating. Generic drugs with an A rating are considered by the FDA to be bioequivalent to the brand name drug. Generic drugs with a B rating are not considered by the FDA to be bioequivalent by in vivo tests and were instead approved by in vitro tests. These drugs are mostly older drugs and only 3 % of generic drugs are currently B rated.
Drug half-life
The time needed for a drug level to be one half of its original concentration is called the elimination half-life. The elimination half-life (t1/2) and the elimination rate constant (K) are intimately connected and form the equation K=0.693/t1/2. This equation allows you to determine the elimination rate constant of a first order reaction if you know a drug’s half-life and conversely determine a drug’s half-life if you know the elimination rate constant. A drug’s half-life often determines how often a drug needs to be administered. Generally, a drug is considered to be at steady state in the body after approximately 5 half-lives.
It is important to remember that drugs often have active metabolites and these metabolites may or may not have similar kinetic properties. The half-life of the original parent drug may or may not be the same as its metabolites.
Volume of distribution
Volume of distribution (VD) is also known as the apparent volume of distribution. A drug’s apparent volume of distribution is a theoretical number that approximates how widely the drug is distributed throughout the body after a drug is given. The VD of any given drug can be affected by several factors including renal failure, dehydration, shock and blood loss. Additionally, the VD of many drugs will vary with age. Drugs that are significantly protein bound and generally stay in the vascular compartment have a VD that is below 10L. Drugs that are not protein bound, lipophilic and move into fatty tissues have a VD that is well over 15,000L. Drugs with a VD between these two numbers distribute out of the vascular compartment, but not entirely into fatty tissue. The following equation can be used to determine VD.
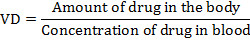
Compartment models
Pharmacokinetic models are categorized as either one-compartment open models or multi-compartment models. A one-compartment open model is used when a drug is given intravenously and makes the assumption that the drug is immediately absorbed and distributed throughout the body. If you were to graph this absorption, it would be a linear line.
A two-compartment open model takes into account that some drugs when administered intravenously, do not absorb into the body in an exactly linear fashion. This model has an initial, brief distribution phase and then a linear elimination phase. This model is only accurate for drugs that are eliminated via a first-order process.
A three-compartment open model works essentially like the two-compartment open model and takes into account that some drugs distribute into deep tissues, which is generally a slow process.
Therapeutic drug monitoring
Therapeutic drug monitoring is a very important aspect of pharmacokinetics. It is important to remember that all drugs have a therapeutic range as opposed to a finite value. However, some medications such as digoxin, theophylline, lithium, aminoglycosides warfarin and many anti-seizure medications have a very narrow therapeutic range. This means that below its therapeutic range, the drug is ineffective and above, it is potentially toxic.
Therapeutic drug monitoring is most importantly focused on drugs that have a narrow therapeutic range. Drugs with a narrow therapeutic range are usually individually dosed and are closely monitored by a patient’s physician and pharmacist. Most drugs in this class have laboratory tests that can be performed to get patient specific drug levels. It is also extremely important to follow these patients closely for potential adverse medication reactions.
Concentration-dependent killing
Aminoglycosides are a class of drugs that have a very narrow therapeutic window and exhibit concentration-dependent bacterial killing. This means that aminoglycosides have the best bacterial killing potential when the concentration of the drug far exceeds the minimum inhibitory concentration (MIC) of the bacteria. Ideally, the peak should be 8-10 times greater than the MIC of the bacteria. Additionally, aminoglycosides are subject to a post-antibiotic effect. This post-antibiotic effect means that the bacteria will not benefit from additional doses of antibiotics for a period of time.
Generally, a target aminoglycoside peak is between 6 and 10 mcg/mL. The lowest effective peak should be used due to the potential of ototoxicity and nephrotoxicity. A target trough of below 1 mcg/mL is ideal.
Time-dependent killing
Vancomycin is a drug that exhibits time-dependent bacterial killing and is generally only used in its intravenous form for gram positive cocci. Antibiotics that kill bacteria in a time-dependent fashion work best when antibiotic concentrations stay about the desired peak for as long as possible. It is best to dose vancomycin so that goal peaks are met and troughs do not drop to zero. Vancomycin is renally cleared and it is very important to renally dose this when needed.
Generally, a target vancomycin peak is between 10 and 30 mcg/mL. Higher troughs tend to have a higher incidence of adverse reactions such as neutropenia and potentially nephrotoxicity. As a general rule, only vancomycin peaks are measure and monitored. Vancomycin troughs can be monitored in critically ill and unstable patients.
Linear kinetics
Linear kinetics is also known as linear biotransformation or first order kinetics. When a drug follows linear kinetics, how fast the drug is metabolized in the body is directly proportional to the amount of the drug given. If you were to plot this on a graph, you would simply get a linear line that has a slope equal to –k/2.303, where k is the first order elimination rate constant. The elimination rate constant and half-life of drugs that follow first order kinetics are independent of the amount of drug given. Most drugs in general follow linear kinetics.
Non-linear kinetics
Non-linear kinetics is also known as non-linear biotransformation or zero order kinetics and in general, few drugs exhibit non-linear kinetics as an exclusive form of metabolism. Drugs such as ethanol, phenytoin and salicylates do exhibit exclusive non-linear kinetics. When drugs such as phenytoin follow non-linear kinetics, how fast the drug is metabolized in the body is not directly proportional to the amount of drug given. Non-linear biotransformation of drugs is most commonly due to these drugs needing to be transported via a protein carrier before biotransformation can occur. There are a finite number of protein carriers and therefore these drugs are dependent on how fast they can pass through these protein carriers.
Phase 1 and 2 biotransformation
Metabolism and biotransformation of drugs can be separated into two components: phase 1 and phase 2. During phase 1 biotransformation, a functional group such as a phenol group or amine group is either attached or uncovered on the main drug compound.
Phase 2 biotransformation is also called phase 2 conjugation. During phase 2 biotransformation, functional groups such as glucuronic acid and other groups are added to the previously exposed functional group. The goal of this conjugation is to make the drug very polar. Polar drugs are in general easier to excrete renally as compared to electrically neutral drugs.
Not all drugs undergo phase 1 and phase 2 biotransformation. Some drugs only undergo phase 2 biotransformation and some drugs do not undergo any biotransformation. These drugs are generally very polar in nature and are easily renally excreted.
Pharmacogenomics and Genetics
Pharmacogenomics
Pharmacogenomics is a generally new scientific field by which genomic techniques are used to measure differences in gene expression of individuals and populations, which might lead to a better understanding of drug response variability. By understanding the underlying regulation of drug metabolism in a given individual can allow for a more individualized therapeutic regimen. Single nucleotide polymorphisms (SNP), are mutations, generally of a coding region of a gene, of a single nucleotide that occur at about a 5% frequency within the population. If these mutations do not change the overall protein sequence or protein function, it does not really matter; however, when these SNPs alter protein function, they can lead to small but significant changes in the phenotype, which can be important in the function and metabolism of a drug.
Desensitization of drug receptors
Desensitization is also known as tolerance and may occur for numerous reasons. One reason is that a receptor becomes oversaturated by a ligand or drug. Receptors will become absorbed into the cells, in which the ligand is removed and the receptor is either degraded or returned to the cellular membrane. This process which is necessary to recycle receptors that have become saturated, it can lead to a down-regulation of drug activity and a down-regulation of therapeutic effects. Some drugs become tolerated faster than others. Morphine is a common drug that becomes well-tolerated or desensitized relatively quickly, and increased doses are needed to reach the same effect. Many tolerances toward a drug are generally reversible.
Pharmacists’ role
Pharmacists will play an important role in pharmacogenetics/pharmacogenomics. Pharmacists can have important roles in researching the effect of important single nucleotide polymorphisms (SNPs) which can regulate drug activity in a certain subset of the population. Pharmacists can have important roles in reading genetic tests, individualizing treatments based on the patients’ genetic makeup, counseling people about their particular genetic polymorphisms so that the patient can understand more about their own healthcare issues. For this pharmacists will need a better understanding of pharmacogenomics in order to be strong leaders in healthcare in the not so distant future.
Genetic drift
Genetic drift is the chance that an allele will mutate, leading to various changes in biological traits but generally does not alter the entire genetics of the population, does not offer any adaptation, and thus is technically separate from natural selection, although the two processes often occur simultaneously. Genetic drift is a common reason for small genetic differences between people and population subsets, such as polymorphisms. Genetic drift also occurs in viruses and other microbial agents, such as influenza, which is important in the variability of the virus that is observed throughout the world and why it is difficult to create an effective vaccine.
SNP of thiopurine methyltransferase
A relatively common SNP causes a defect in the thiopurine methyltransferase (TMPT). The enzyme is important in methylation of thiopurine compounds. It also has important roles in regulating chemotherapy drugs such as 6-methylcaptoporurine, azathioprine, and 6-thioguanine, all of which have important immunosuppressive qualities. People with a mutated TMPT often cannot metabolize these drugs properly which can often lead to severe adverse reactions such as severe bone marrow toxicity, which occurs when bone marrow is severely depressed. This suppression of the bone marrow allows for acute infections to take hold and possibly be fatal to the patient. New tests are now available for TMPT defects. It is presently used to titer doses for individual people. People without the defect get the normal dose of the immunosuppressants, while people with the defect will get a much lower dose and still show therapeutic effects.
Pharmacogenetic testing challenges
Pharmacogenetic testing can be very difficult and overwhelming to perform, particularly since the amount of information out there is immense. Ideally it would be nice to be able to test the complete genome for any possible SNPs, receiving a complete view of every individual’s genetic makeup in order to efficiently and effectively individualize therapy. However, in reality, this is not feasible, as it would take a massive amount of time and resources to do this. In reality it would be more feasible to test particular gene SNPs, thus keeping the pharmacogenetic tests manageable. This could unfortunately lead to the neglect of other prominent SNPs. It is also important to understand and study the mechanistic relationship between genotype and phenotype further. An understanding of the link between observed genetic SNPs and mutated phenotypes is necessary before truly effective testing can be performed. This type of testing is important, however, and even as limited as it is now, it can save countless lives.
Altering metabolism of codeine and Bidil
A small subset of the Caucasian population has a mutation in the CYP2D6 enzyme. CYP2D6 is an important metabolic enzyme. It has roles in metabolizing the opioid drug codeine. To be effective, codeine must be metabolized into morphine. People lacking a proper functioning CYP2D6 enzyme, cannot metabolize the drug into morphine, thus codeine has no effect. Another example of population differences in relationship to drug therapies include isosorbide dinitrate and hydralazine (Bidil) was a drug that was originally denied by the FDA because it didn’t show an effective treatment against heart disease in the general population, but in later tests showed a significant effect in a subset of the African American population. The genetic basis for this drug therapy has not been fully elucidated.
Linkage disequilibrium
Linkage disequilibrium is a term used in genetics to describe the association of alleles at two different loci (fixed genetic position of a chromosome) often on different chromosomes. Linkage disequilibrium is used in population genetics to measure the frequency of haplotype formation and genetic changes which deviates from the frequency if mutations were simply random. It is thought that this process is evolutionarily conserved, allowing for greater genetic variability within a population, and also leads to the formation of SNPs. Many factors can regulate linkage disequilibrium including natural selection, genetic drift, gene conversion, rates of genetic recombination, as well as rates of mutation.
Diminished metabolizers and ultra-metabolizers
Often times SNPs will cause one of two types of changes in drug metabolizing; these include diminished metabolizers and ultra-metabolizers. Diminished metabolizers, often caused by mutations in the metabolic pathway which generally leads to increased bioavailability of the drug, increased drug activity, and increased toxicity if the patient took an active compound. If a poor-metabolizer took a pro-drug however, this could lead to a diminished response and decreased bioavailability. Ultra-metabolizers have increased levels of metabolism, which can lead to a decreased bioavailability, and decreased effectiveness of the drug if the patient took an active drug. If they are taking a pro-drug, however, this could increase the bioavailability of the active component and lead to a toxic response.
Polymorphisms of MDR1, 2AR, and SUR
24% of the population has a SNP in the multi-drug resistance gene 1 (MDR1), a drug transporter, which can lead to an increased amount of plasma levels of Digoxin, which can lead to increased bioavailability and increased toxicity of the drug. 37 % of the population has a SNP in the Beta-2 adrenergic (2AR) receptors which can weaken the response of beta-2 agonists such as Albuterol. 3% of the population has a SNP in the Sulphonylurea receptor gene (SUR) which can lead to decreased insulin response and can decrease the activity of Tolbutamide.
Gender differences in drug metabolism
There is increasing evidence that gender differences exist in relationship to drug metabolism, although these differences are not entirely understood. Certain drugs for example have faster metabolism in the different sexes. Propranolol was metabolized at a rate 50% faster in men than in women through oxidative metabolism. Other drugs such as chlordiazepoxide and lidocaine are also metabolized faster in men than women. Other drugs are metabolized faster in women than men such as caffeine, diazepam, and acetaminophen. These differences can be in part due to differences in phase I and phase II metabolism, such as differences in oxidation reactions and cytochrome p450 enzymes.
Glucose 6-phosphate dehydrogenase deficiencies
Deficiencies can occur in glucose 6-phosphate dehydrogenase which can lead to several blood problems such as hemolytic anemia. Historically this disorder has gone seemingly unnoticed until the ingestion of uncooked fava beans, which causes the victim to fall very ill. Blood cells which show a sharp decline in 6-phosphate dehydrogenase undergo hemolysis in response to uncooked fava beans as well as a few particular drugs. The exact mechanism is not well understood. People with this deficiency should not be given anti- malarial drugs, sulfonamides, nitrofurans, acetaminophen, acetylsalicylic acid or vitamin K. This deficiency is more common in males, as it is an X-linked sex trait; however, homozygous females are somewhat common in comparison to other sex linked disorders.
Polymorphisms of UDP glucuronosyltransferase SNPs
UDP glucuronosyltransferase (UGT) 1A6 is an important component of the glucoronidation metabolism pathways found in the liver. Glucoronidation metabolism is important in the detoxification of phase I drug metabolites. Polymorphisms of the UGT1A6 gene can have major effects on drug metabolism. It has been shown that UGT1A6 can lead to problems in metabolism of phenolic drugs. Heterozygous UGT1A6 SNPs have moderate decreased activity of many drugs, due to increased metabolism. People who possess homozygous SNPs of UGT1A6 have a higher level of glucoronidation and decreased activity of many drugs including acetaminophen, morphine, beta-blockers, and cyclosporine A. SNPs in UGT1A1 were shown to lead to increased adverse drug reactions in relationship to irinotecan. Additionally, UGT1A1 SNPs have been connected to disorders including Gilbert’s syndrome and Crigler-Najjar Syndrome. Gilbert’s Syndrome is a heritable disease that leads to chronic decreased metabolism of bilirubin, leading to a mild decrease in clearance of the protein. Crigler-Najjar Syndrome is a more severe disease involved in decreased bilirubin metabolism, which is chronic and can be fatal if untreated.
N-acetyltransferase polymorphisms
There are two forms of N-acetyltransferase (NAT1 and NAT2), both of which can have important polymorphisms. N-acetyltransferase has important roles in metabolism, which transfers the acetyl groups to arylamine compounds. Acetylation is an important step in both synthesis and breakdown of numerous biological components. More is known however about the clinical relevance of NAT2 polymorphisms. NAT2 polymorphisms can lead to adverse drug reactions to such drugs as isoniazid, hydralazine, procainamide, sulphasalazine. People administered with isoniazid who have a NAT2 polymorphism suffer from peripheral neuropathy. Other adverse reactions include lupus erythematosus with administration of hydralazine or procainamide and hemolysis in response to sulphasalazine.
Sterile and Nonsterile Compounding
Compounding terms
Admixture—IV drugs that are sterilely combined together in one IV bag before administration.
Aseptic technique—performing procedures in a sterile environment to minimize contamination.
Controlled Area (buffer area)—the area where the laminar flow workbench is located within the pharmacy. This area should remain as clean as possible to reduce contamination chances.
Critical site—any space that can lead to a possible contamination between a sterile product and the outside environment. Must be tightly controlled and regulated.
Hypertonic—solution that has higher levels of solutes than red blood cells, which will lead to shrinking of the cells.
Hypotonic—opposite to hypertonic, can lead to bursting of red blood cells.
Isotonic—solutions that are similar to body fluids, similar osmotic and diffusive pressures which should not lead to stress on red blood cells and other cell types.
Sterilization techniques
Filtration—uses a filter with a certain pore size such as 0.2 micron, in which anything above 0.2 microns will not go through the filter. Generally, compounded products should be filter sterilized with a 0.2 micron filter; however, to avoid clogging of the filter, a larger pore size should be used first to remove bigger particles. Two types of filters are generally used, hydrophilic filters for aqueous solutions and hydrophobic filters for gases and volatile solvents.
Heat sterilization is another technique used by compounding pharmacies and research labs. Autoclaving uses a mixture of heat, moisture and pressure to kill microbial agents and sterilize solutions and labware. It is usually quick, yet very effective. Dry sterilization uses an oven at very high temperatures and filtered air to sterilize products. Dry sterilization uses hotter temperatures for longer periods of time than an autoclave and is used to remove impurities (pyrogens) from lab and compounding equipment.
Quality compounding guidelines
There are important quality control guidelines that should be followed when compounding. These guidelines include maintaining the utmost care when compounding any drug and not cutting any corners. It important to maintain accurate measurements, processing procedures, packaging, record-keeping and properly assigned beyond-use-dates. Beyond-use-dates are expiration dates assigned by the pharmacist that are based on drug degradation. A pharmacist, when deciding on a beyond-use-date, needs to strike a balance long enough for the drug to be used by the patient according to the dosing regimen on the prescription, but not too long that the drug will degrade and become useless for therapeutic treatment.
Tablets
A tablet is a solid formulation of a drug made for the purpose of easier storage and administration. Effervescence tablets are dissolved in water before they are taken. Effervescence occurs when carbon dioxide is released through the reaction of sodium bicarbonate in water, which helps dissolve the tablet and release the drug into the solution. Chewable tablets are used for a more rapid absorption of a drug. The drug is released through chewing and interaction with saliva. Buccal and sublingual tablets are dissolved in the presence of saliva. These type of drugs are preferred if avoidance of the first-pass effect is desired. Time release tablets can also be called sustained released tablets, which are formulations that have several doses of a drug, though each dose is not released into the system until a certain time has passed. They can potentially reduce adverse reactions and improve adherence of a drug. Coated tablets are created to improve taste and to prevent premature dissolving once swallowed. Enteric-coated drugs for example resist the stomach acids and dissolve in the intestines.
A tablet is a mixture of a drug and inactive carrier/ ingredients that is formulated into a solid, generally for easy oral consumption and absorption into the body, often through the gastrointestinal tract. Tablets are formulated with various agents including diluents, adsorbents, moistening agents, binding agents, glidants, lubricants and disintegrating agents. Diluent agents are also known as bulking agents such as sucrose of NaCl, which is used with low-dose drugs to bring the weight of the tablet to at least 50 mg, for easier handling and use. Adsorbents are agents that can bind and sequester fluids keeping them dry, in granules before being compressed into a tablet. Absorbents include silica. Moistening agents are used to keep compounds during a wet granulation process. Moistening agents include water and isopropanol. Binding agents are used to adhere moist granulations during tablet formation. Binding agents include starch, cellulose, and polyvinylpyrrolidone. Glidants are compounds use to improve friction and flow of granulated components of a tablet. Glidants include silica, starch and talc powder. Lubricants are also used, mainly during the manufacturing process so that the tablets do not stick or remain in the manufacturing machines, thus the tablets do not break.
Emulsions
An emulsion consists of at least two liquids that will not mix with each other such as oil and water. Instead one of the liquid phases (dispersed phase) (i.e. oil) will be diffused throughout the second liquid phase (continuous phase) (i.e. water) as droplets or globules. This mixture is stabilized by emulsifying agents. The two most common types of emulsions are oil-in water (o/w) or water-in-oil (w/o). O/w emulsions generally have oil globules dissolved into an aqueous phase. W/o emulsions have water globules dissolved into an oil phase. Emulsions are often used as lotions, ointments and creams. Common o/w emulsifying agents include sodium lauryl sulfate, sodium oleate and glyceryl monosterate. Common w/o emulsifying agents include calcium palmitate, sorbitan esters, cholesterol and wool fat products.
Emulsions can become unstable by creaming and sedimentation, breaking, coalescence, aggregation, phase inversion and through microorganism growth. Creaming occurs in response to an upward movement of particles (globules) in the dispersed phase in relation to the continuous phase (i.e. oil droplets moving upward through the aqueous phase). Sedimentation is the opposite of creaming, when the particles are globules move downward through the continuous phase. Both are reversible by remixing the emulsion compound but can lead to other problems in emulsion instability. If during sedimentation or creaming, the protective film that surrounds the dispersed globules breaks, this can lead to breaking of the emulsion, causing the globules to coalesce into larger globular particles or aggregate into larger clumps. Phase inversion occurs when an emulsion goes from a w/o emulsion to an o/w emulsion or o/w emulsion to a w/o emulsion. Changes in electrolytes and the addition of different emulsifying agents can lead to this effect. Also, microorganisms can grow on emulsion destroying emulsifying agents. Preservatives are usually added to prevent such a thing to occur.
Shelf-life
Drugs have a finite period of time before they degrade and are no longer effective therapeutically. This period of time, which begins as soon as the drug is manufactured and stored, is known as the shelf-life of a drug. Shelf-life of a drug or dosage form of a drug can be determined by using the Arrhenius equation: log (k2/k1) = (Ea (T2-T1))/2.303RT2T1. k2 and k1 are the rates of the reaction at the absolute temperatures which are described as T2 & T1. R= the gas constant. Ea is the activation energy of the experiment. This reaction can give a best estimate of the amount of time that a drug will be effective before full degradation occurs.
Dosage forms of drugs
Solutions are homogenous mixtures of one or more substances in a solvent such as water. There are different types of solutions, including syrups, elixirs, and tinctures. A syrup consists of sugar, usually sucrose, or other flavoring agents dissolved in water along with the drug of choice. An elixir consists of a sweetened solution of water and ethanol, along with the drug or drugs of choice. A spirit is generally considered a solution of alcohol and water that is mixed with volatile and pungent compounds and drugs. Tinctures are generally vegetable based extracts formed in either alcohol or alcohol/water solutions. Solutions are often taken orally. If kept sterile, solutions can also be used for injectable drugs such as antibiotics, pain medication, etc.
Type A prescription balances
Type A (type III) prescription balances are an important component of a compounding pharmacy and are used to accurately weigh and measure reagents in order to create a functional and safe therapeutic drug. Accuracy of a type A prescription balance must be kept in good working condition, and its sensitivity and accuracy should be determined periodically. The United Standards Procedure (USP) regulates four specific tests to determine the sensitivity of the balance: the sensitivity determination test, the arm-ratio test, shift test and the rider graduate-beam test. The sensitivity determination test requires a 6 mg weight to determine the sensitivity of the balance up to a 6 mg. The arm-ratio test is used to detect inaccuracies between the lengths of the arms of the balance. The shift test is used to determine if the arms and the levels of the balance have been shifted out of place, a good test to determine misuse of the balance. The graduate-beam test is used to measure inaccuracies of the weigh-beam and rider of the balance.
HEPA filters
HEPA stands for High Efficiency Particulate Air filter or High Efficiency Particulate Arresting filter. It is an air filter that is important in compounding hoods to maintain sterility of the drugs and other compounding reagents needed to compound the drug. The HEPA filter acts as a blockade, made up of meshed fibers spaced no more than 3 µm apart, thus removing anything larger than 3 µm. Additionally other smaller compounds will often stick to the meshed fibers, rather than crossing through the filter. Using a fast airstream across the filter can greatly decrease the diffusion rate of particulates in the air, to an area of 0.1 µm, easily inhibiting bacteria and viral contamination. Efficiency of HEPA filters should be tested often and replaced when efficiency drops in order to maintain the integrity of the compounding pharmacy.
Measuring laminar flow efficiency
Smoke test—by creating a “smoky” atmosphere. This can be performed a number of ways, including putting dry ice in water and observing the dissipation of “smoke” in the laminar hood, to measure airflow, to determine that the hood is working properly.
DOP Test— using aerosol DiOctyl Phthalate (DOP). The DOP test can be used to measure particulate penetration of the HEPA filter in the laminar hood.
Microbial Test—with the use of a device known as a microbial sampler, can be used to sample air quality of the laminar hood to measure levels of microorganisms and can also be used to measure other levels of possible contaminates.
Ethanol and ultraviolet sterilization
Ethanol Sterilization—70% ethanol is considered to be the most effective concentration of ethanol for sterilization techniques. A greater percentage of ethanol does not provide greater sterilization, and thus, it is more cost-effective to use 70% ethanol. Ethanol kills bacteria and other organisms by denaturing proteins and disrupting the cell wall/cell membrane of cells. Additionally, it is thought that 95% – 100% ethanol evaporates so rapidly that it does not have a chance to fully kill bacteria and other microbes.
Ultraviolet (UV) Sterilization—many hoods are connected with ultraviolet lights which are used for sterilization. At a wavelength of approximately 250 nm, the UV light can lead to DNA breaks which can lead to thymine dimers in DNA, inhibiting replication, transcription and translation of the microbial organisms, leading to the death of the microbial cells.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


