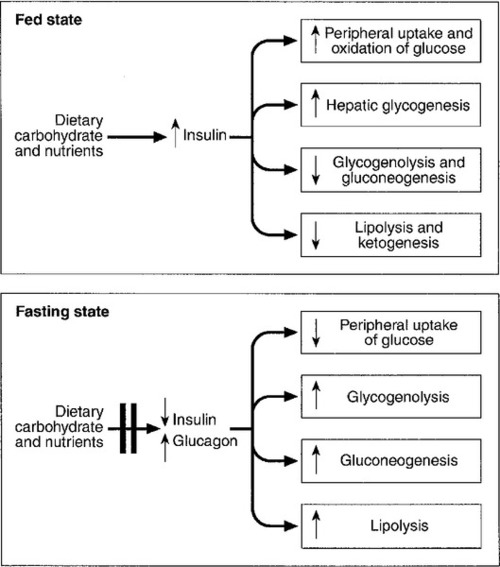
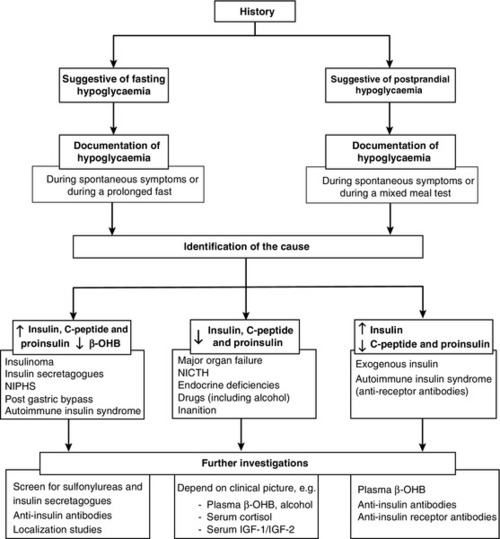
CHAPTER 17 CHAPTER OUTLINE GLUCOSE HOMOEOSTASIS IN THE FED AND THE POSTABSORPTIVE STATES The neuroendocrine response to hypoglycaemia CLASSIFICATION OF HYPOGLYCAEMIC DISORDERS PRACTICAL APPROACH TO THE INVESTIGATION OF HYPOGLYCAEMIA Evaluation of hypoglycaemia in persons without diabetes mellitus Investigation of hypoglycaemia Evaluation of hypoglycaemia in patients with diabetes mellitus EMERGENCY TREATMENT OF HYPOGLYCAEMIA Surreptitious administration of hypoglycaemic agents (factitious or felonious hypoglycaemia) Islet cell tumours (insulinoma) Non-insulinoma pancreatogenous hypoglycaemia syndrome (NIPHS) Non-islet cell tumour hypoglycaemia (NICTH) Hypoglycaemia associated with renal impairment Hypoglycaemia associated with liver disease Hypoglycaemia due to endocrine deficiencies Hypoglycaemia due to deficient energy intake Exercise-related hypoglycaemia Postprandial (reactive) hypoglycaemia In healthy subjects, the blood glucose concentration is maintained within relatively narrow limits through a tightly controlled balance between glucose production and glucose utilization. Fundamentally, glucose is derived either from dietary intake (in the fed state) or from glycogenolysis and gluconeogenesis (in the fasting or postabsorptive state). It is metabolized by oxidation or stored either in the form of glycogen or through conversion to fat (Fig. 17.1). FIGURE 17.1 Glucose homoeostasis in the fed and fasting states. Plasma glucose regulation is a complex process involving both insulin-dependent and insulin-independent mechanisms. This regulation is a multi-organ process involving the gastrointestinal tract, pancreas, liver, muscles, adipose tissue, brain and kidneys. In the gastrointestinal tract, incretin hormones (see below), secreted in response to meals, promote glucose-mediated insulin secretion and suppress glucagon production. In the liver, insulin regulates blood glucose concentrations by suppressing hepatic glucose output and increasing postprandial glucose storage in the form of glycogen. In muscle and adipose tissue, insulin binding to insulin receptors leads to increased expression of glucose transporter 4 (GLUT4) molecules in the cell membrane, facilitating glucose uptake. Insulin-independent mechanisms that contribute to glucose regulation are located in different organs, but mainly in the gastrointestinal tract and the kidneys. Sodium–glucose cotransporters (SGLT), such as SGLT1 in the gastrointestinal tract and SGLT2 in kidney, are important mediators of insulin-independent glucose transport across cell membranes. In response to nutrient ingestion, glucagon-like peptide (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are released from enteroendocrine cells. Glucagon-like peptide-1 enhances glucose-dependent insulin secretion and inhibits glucagon secretion. Glucose-dependent insulinotropic peptide also stimulates glucose-dependent insulin release, and the combined actions of GLP-1 and GIP account for up to 60% of the overall postprandial insulin secretion. The increase in glucose and insulin concentrations results in stimulation of glycogen synthase and inhibition of glycogen phosphorylase, causing a net increase in hepatic glycogen. Approximately 50% of an oral glucose load is taken up by the liver and 50% by peripheral insulin-sensitive tissues. Three to four hours following a meal, glucose and insulin concentrations decline and the liver switches from net glucose uptake to net glucose release. Hepatic glucose release continues to increase over the next several hours until it equals glucose utilization. Initially, about 75% of glucose production is attributable to glycogenolysis and 25% to gluconeogenesis, but as hepatic glycogen stores decline, the proportion from gluconeogenesis increases steadily. During the early postabsorptive phase, glucose utilization continues at the rate of 0.10–0.14 mmol/kg per min, of which 40–50% is due to obligatory uptake by the brain and other non-insulin-dependent tissues. With prolonged fasting, the decline in plasma insulin concentration is accompanied by increases in plasma free fatty acid and ketone body concentrations. These can be used as alternative fuels, thereby decreasing the need for glucose. The definition of hypoglycaemia is somewhat arbitrary since the glycaemic threshold at which symptoms occur differs between individuals, and depends on the age of the patient and the prevailing plasma glucose concentration before the hypoglycaemic episode. A venous plasma glucose concentration <3.0 mmol/L, regardless of the presence or absence of symptoms, has been recommended as the biochemical definition of hypoglycaemia by The Endocrine Society in the USA. However, many apparently healthy people, especially women, may have plasma glucose concentrations <3.0 mmol/L during a prolonged fast without experiencing any symptoms. Conversely, diabetic patients may develop hypoglycaemic symptoms at higher plasma glucose concentrations. Therefore, the diagnosis of pathological hypoglycaemia necessitates the demonstration of Whipple’s triad. This consists of: (1) either symptoms or signs, or both, consistent with hypoglycaemia in the presence of (2) a low plasma glucose concentration, with (3) the relief of symptoms or signs after the plasma glucose concentration is raised. Under normal metabolic conditions, in the unstressed state, the central nervous system (CNS) is wholly dependent upon glucose as its primary source of energy. The brain requires approximately 150 g of glucose each day, which must be continuously available. The presence of hypoglycaemia triggers a complex and well-coordinated metabolic process aimed at limiting and preventing a further fall in blood glucose concentration. The extent and duration of hypoglycaemia are primary factors in determining the magnitude of this counter-regulatory response, but other factors, such as age, sex, rate of fall of blood glucose and insulin per se have also been shown to influence the response. In healthy subjects, the initial response to prevent a decline in blood glucose concentration is a reduction in insulin secretion, which begins while the plasma glucose concentration is falling but still within the physiological range. This is followed by an increase in glucagon and adrenaline (epinephrine) secretion as the glucose concentration declines further. Glucagon, secreted by pancreatic α-cells, rapidly raises plasma glucose concentration by stimulating hepatic glucose production via glycogenolysis and gluconeogenesis. The activation of the autonomic nervous system, mainly the sympathetic, increases the concentrations of noradrenaline (norepinephrine) at nerve terminals and adrenaline in the circulation. Adrenaline acts on α- and β-adrenergic receptors at several end organs and results in a more sustained increase in blood glucose concentration. It reduces insulin secretion from the pancreatic islets, increases glycogenolysis and gluconeogenesis in liver, increases glycolysis in muscle and increases lipolysis in adipose tissue. Hepatic glycogen stores provide glucose, particularly for the brain, whereas the mobilization of fatty acids from fat depots provides energy for tissues that can utilize fatty acids as their basic fuel, for example skeletal and cardiac muscles, renal cortex and liver, thus sparing glucose for use by the CNS. As blood glucose concentration declines further, hypothalamic sensing of hypoglycaemia results in increased secretion of growth hormone (GH) and adrenocorticotropic hormone (ACTH) and, consequently, cortisol. The increase in GH and cortisol secretion induces metabolic changes over longer periods of time by stimulating lipolysis in adipose tissue and ketogenesis and gluconeogenesis in the liver. The glycaemic thresholds for the activation of these responses are higher than those for the development of symptoms or the impairment of cognitive function. With falling plasma glucose concentrations, the typical sequence of responses (and their glycaemic thresholds) is as follows: decrease in endogenous insulin secretion (4.4 mmol/L); increase in glucagon and adrenaline (3.8 mmol/L); increase in GH secretion (3.7 mmol/L); increase in cortisol secretion (3.2 mmol/L); development of symptoms (3.0 mmol/L), and impairment of cognitive function (2.5 mmol/L). The symptoms of hypoglycaemia are non-specific and vary depending on the degree of hypoglycaemia, the rapidity of the decline in blood glucose, the age of the patient, whether the hypoglycaemia is clinical or experimental, and on patients’ differing perceptions of symptoms. The symptoms differ from one person to another, but are usually consistent from episode to episode for any one person. In some patients, symptoms are mainly due to activation of the sympathetic nervous system (neurogenic or autonomic), but in others only symptoms due to brain glucose deprivation per se may be observed. All the symptoms of hypoglycaemia, however, reflect the effects of glucose deprivation on the CNS (neuroglycopenia). This is characterized by sweating, anxiety, hunger, tremor, palpitations and weakness (Box 17.1). These symptoms result mainly from activation of the sympathetic nervous system and characteristically occur when there has been a rapid decline in blood glucose concentration. It may occur in patients with diabetes owing to excessive absorption of exogenous insulin, either from overtreatment or from rapid mobilization from the injection site during exercise. In non-diabetic subjects, reactive hypersecretion of insulin may be responsible, for example after Roux-en-Y gastric bypass for obesity. Awareness of hypoglycaemia is mainly the result of the perception of neurogenic symptoms which may be lost in patients with type 1 diabetes mellitus (hypoglycaemia unawareness). The symptoms of acute neuroglycopenia may mimic those of anxiety attacks or hyperventilation. Symptoms range from a generalized decrease in spontaneous activity, poor concentration, episodic disorientation, confusion, incoordination, slurred speech and behavioural changes, to seizures, focal neurological signs and coma (Box 17.1). Patients with subacute neuroglycopenic symptoms have been misdiagnosed as having epilepsy, transient ischaemic attacks, psychosis, hysteria, chronic nervous exhaustion, personality disorder or inebriation. Chronic neuroglycopenia is very rare and may occur in patients with insulin-secreting tumours unsuspected for years. Symptoms include insidious progressive mental disturbance that may resemble personality disorders, schizophrenia, paranoid psychosis, depression or dementia (Box 17.1). Several classifications of hypoglycaemia are possible, for example aetiological, ketotic or non-ketotic, a classification based on the timing of symptoms in relation to meals (fasting or postprandial) or whether hypoglycaemia occurs in a healthy person or one who has an illness. The usefulness of classifying hypoglycaemic disorders based on the timing of symptoms in relation to meals has been questioned because some conditions that are associated with fasting hypoglycaemia, such as insulinoma, may also produce symptoms postprandially, and post-gastric bypass patients, who typically have postprandial hypoglycaemia, may have symptoms when fasting. Also, patients with factitious hypoglycaemia may have symptoms that have no clear relationship to food intake. In view of this, a classification which is based on the clinical characteristics, i.e. on whether hypoglycaemia occurs in a seemingly well individual or an ill or medicated individual, is more useful (Box 17.2). The direction of investigation will depend essentially on the clinical presentation and whether the patient appears ill or not. Hospitalized patients are often severely ill with multisystem disease. In such cases, the underlying illnesses that can produce episodes of hypoglycaemia, such as severe renal or hepatic disease, congestive cardiac failure, sepsis and anorexia nervosa, are usually obvious clinically. A review of all medications is essential since drugs (see below) are a common cause of hypoglycaemia in hospitalized patients, particularly in the presence of renal impairment. Non-islet cell tumours causing hypoglycaemia are usually, though not invariably, large mesenchymal tumours that are clinically apparent. In these cases, confirmation of the suspected mechanism of the hypoglycaemia may be sought by measuring plasma insulin, C-peptide, proinsulin and β-hydroxybutyrate (β-OHB) concentrations during an episode of hypoglycaemia. Endocrine deficiencies such as hypopituitarism and adrenocortical insufficiency should be sought and excluded by appropriate investigations if necessary. In a seemingly well individual, with no obvious coexisting disease, the direction and extent of evaluation will depend on the clinical presentation. A thorough and detailed history of the symptoms (frequency, type, relationship to meals and exercise) is therefore essential. If symptoms are relieved by food ingestion, it is important to enquire about the type of food and speed of recovery. All medications should be inspected to exclude a prescribing or dispensing error, and the possibility of surreptitiously induced hypoglycaemia should always be considered, especially in healthcare professionals and relatives and carers of diabetic patients. Assessment of alcohol intake and pattern of drinking is important, since alcohol can cause both fasting and reactive hypoglycaemia. A history of previous gastric surgery or Roux-en-Y gastric bypass surgery for obesity and a family history of multiple endocrine neoplasia type 1 (MEN 1) should be sought. Symptoms in individuals of Japanese or Korean ethnicity may point to autoimmune hypoglycaemia. The aims of the investigation are, first, to demonstrate that hypoglycaemia is the cause of the symptoms and, second, to identify the cause of the hypoglycaemia (Fig. 17.2). FIGURE 17.2 A practical approach to the investigation and diagnosis of hypoglycaemia (β-OHB, β-hydroxybutyrate; NIPHS, non-insulinoma pancreatogenous hypoglycaemia syndrome; NICTH, non-islet cell tumour hypoglycaemia; IGF-1 and IGF-2, insulin-like growth factors 1 and 2). Measurement of the blood glucose concentration (and collection of a suitable blood specimen for subsequent measurement of plasma insulin, C-peptide, proinsulin and β-OHB concentrations) during spontaneous symptoms, and before glucose is given, is without doubt the best test for the diagnosis of spontaneous hypoglycaemia. The most practical way to obtain an accurate glucose concentration is to measure glucose immediately in whole blood or to separate plasma from cells within 30 min of collection, even if the specimen is collected in a tube that contains sodium fluoride. It is well recognized that the rates of decrease of glucose in the first hour after specimen collection in tubes with or without fluoride are virtually identical. The reduction in glucose after 2 h in a fluoride tube can sometimes exceed 0.5 mmol/L leading to a falsely low blood glucose concentration or ‘pseudohypoglycaemia’. In addition, although arteriovenous plasma glucose concentration differences are negligible in the fasting state, antecubital venous plasma glucose concentrations are significantly lower than arterial glucose concentrations after a glucose load owing to glucose extraction across the forearm. Therefore, arterialized blood collected from a vein at the back of the hand, which has been warmed by a heat pad, is preferred. Often, however, patients referred for a medical opinion are asymptomatic when seen in the outpatient clinic, and measuring their blood glucose concentration at such time is usually unhelpful. In this situation, reproducing the circumstances which may lead to hypoglycaemia should be attempted. In patients with a history suggestive of fasting hypoglycaemia, a prolonged fast test should be performed, whereas in patients who only experience symptoms of hypoglycaemia within a few hours of having a meal, a mixed meal test should be performed (see provocation tests, below). The prolonged fast (up to 72 h) is the single most useful investigation employed in the evaluation of patients with suspected spontaneous hypoglycaemia. The aim of the investigation is to demonstrate spontaneous hypoglycaemia in the presence of neuroglycopenic symptoms and the resolution of the symptoms when the plasma glucose concentration is raised to within normal limits. During a prolonged fast, normal subjects rarely develop plasma glucose concentrations <3.0 mmol/L and almost never develop neuroglycopenic symptoms. The fast must be conducted in hospital under strict medical supervision. Before initiation of the fast, a simple assessment of coordination, recent memory and calculations (e.g. counting down in sevens) should be performed as a baseline. The time for initiating the fast is determined depending on a reasonable estimate of the patient’s likely tolerance for fasting. If fasting starts after the evening meal or at midnight, the majority of patients with insulinoma will develop hypoglycaemia by the middle of the following day, when adequate staff and laboratory facilities are available. Patients should have an intravenous indwelling catheter, and an ampoule of 50% glucose solution should be readily available. During the test, the patient is allowed water and non-caloric beverages and should undertake brisk exercise under supervision. Blood, for glucose, insulin, C-peptide, proinsulin and β-OHB measurements, is collected at the beginning of the test, and every 4–6 h thereafter. Plasma glucose should be determined in the laboratory as soon as possible after collection, and plasma or serum should be stored at –20 °C for subsequent analysis of insulin, C-peptide, proinsulin and β-OHB if required. Bedside glucose meters can be used to monitor the patient’s blood glucose concentration frequently, but low values, especially in the absence of symptoms of hypoglycaemia, should always be confirmed in the laboratory. When the plasma glucose has fallen to <3.0 mmol/L, blood should be collected more frequently and the patient should be assessed for neuroglycopenia by repeating the same tests performed at the initiation of the fast. If neuroglycopenic symptoms develop and blood glucose is <3.0 mmol/L, several specimens should be obtained for plasma glucose, insulin, C-peptide, proinsulin and β-OHB measurements before any remedial action, such as glucose administration, is taken. At the end of the fast, a sample should be collected to screen for hypoglycaemic agents and to measure insulin antibodies. A glucagon test (1.0 mg intravenously) has also been recommended (see below). The criteria to stop the test before 72 h are: (1) Whipple’s triad has been observed; (2) plasma glucose concentration <3.0 mmol/L in a patient who had previously experienced Whipple’s triad, and (3) plasma β-OHB >2.7 mmol/L. The measurement of β-OHB concentrations in real-time during the fast is quite useful because a progressive rise in plasma β-OHB concentrations indicates indirectly that the circulating insulin concentration has been suppressed and that the fast can be terminated. However, blood should always be collected in this situation to confirm suppressed plasma insulin, proinsulin and C-peptide concentrations. In patients with insulinoma, about 40% develop hypoglycaemia within 12 h, 75% within 24 h, 95% by 48 h and 99% by 72 h. This test serves as a supplemental study in the diagnosis of hypoglycaemic disorders when results from the prolonged fast are inconclusive. The rationale is that, in patients with insulinoma, there is a greater increase in plasma glucose after intravenous glucagon due to the lesser depletion of hepatic glycogen during the fast as a consequence of the higher insulin secretion. The test is performed at the termination of a prolonged fast test, either at the time of occurrence of symptomatic hypoglycaemia with concomitant plasma glucose concentration <3.0 mmol/L, or after 72 h of fast. Glucagon 1 mg is injected intravenously over 2 min and plasma glucose is measured at baseline, 10, 20 and 30 min after the glucagon injection. An increase in plasma glucose concentration of ≥1.4 mmol/L after intravenous glucagon indicates mediation of the hypoglycaemia by insulin. A disadvantage of this test is the danger of causing severe hypoglycemia after 90–180 min. This test is used to investigate patients who experience only postprandial symptoms. The meal has not been standardized but, ideally, a meal similar in composition to the meal that usually provokes symptoms should be used. Plasma glucose concentration is monitored every 30 min for 5 h and also at any time while the patient is symptomatic. Capillary or arterialized blood (see above) should be used, since postprandial venous plasma glucose concentrations are about 10% lower than arterial concentrations owing to extraction of glucose by muscle. Samples should also be collected every 30 min for insulin, C-peptide and proinsulin measurement but should only be analysed if they have been collected at the time when plasma glucose concentration was <3.0 mmol/L and before administering carbohydrates. Patients should be observed for symptoms, and the timing of these symptoms in relation to the meal, and to the blood glucose concentrations, should be documented. If Whipple’s triad is demonstrated, a sample for the measurement of oral hypoglycaemic agents and insulin antibodies should be collected. Patients who develop neuroglycopenic symptoms during hypoglycaemia, but not at other times during the test, may be considered to have postprandial hypoglycaemia. However, in all patients with a positive mixed meal test, additional testing, including a supervised 72 h fast, is recommended. Postprandial hypoglycaemia without fasting hypoglycaemia has been documented in some patients with insulinoma and in patients with non-insulinoma pancreatogenous hypoglycaemia (see p. 343). The diagnosis of postprandial hypoglycaemia should not be made on the basis of an oral glucose tolerance test (OGTT). At least 10% of ‘normal’ subjects may have plasma glucose nadirs of <2.6 mmol/L during an OGTT and 2.5% may have values <2.2 mmol/L. Moreover, symptoms during OGTT are often unrelated to the level of plasma glucose nadir or to the rate of decline of the glucose concentration. Although insulin and C-peptide are secreted in equimolar amounts by the β-Cells of the pancreas, the metabolic clearance of insulin is much more rapid than that of C-peptide. Therefore, C-peptide has a longer half-life and is present in peripheral blood in higher molar concentrations than insulin, making it less prone to marked fluctuations. Consequently, the measurement of plasma C-peptide concentrations may be more reliable as an indication of endogenous insulin production. However, as C-peptide is cleared by the kidneys, raised concentrations may occur in renal impairment. Intact proinsulin undergoes enzymatic processing leading to production of des-31,32-proinsulin and des-64,65-proinsulin and then to insulin and C-peptide. Some proinsulin assays specifically measure intact proinsulin whereas others measure ‘total proinsulin’, i.e. intact and other molecules such as des-31,32-proinsulin. Therefore, proinsulin results depend strongly on the assay used and should be interpreted with caution. In islet cell tumours, the amount of circulating proinsulin is increased and, occasionally, tumours may secrete mainly or exclusively proinsulin. A plasma proinsulin concentration ≥5.0 pmol/L, when the plasma glucose concentration is <2.5 mmol/L during a 72 h fast test, represents the best criterion for the diagnosis of endogenous hyperinsulinaemia with 100% specificity and sensitivity. In view of this, the Endocrine Society has now recommended the measurement of plasma proinsulin as a first-line test, in addition to insulin and C-peptide. Insulin and C-peptide can be measured in either plasma (lithium heparin) or serum, as long as the serum or plasma is separated within 15 min of collection and frozen immediately following separation. Plasma insulin concentrations can be artefactually modified by heterophilic antibodies, endogenous anti-insulin antibodies or haemolysis, as red blood cells contain an insulin-degrading enzyme, which may lead to underestimation of the actual insulin concentrations in haemolysed samples. The measurement of plasma insulin, C-peptide and proinsulin concentrations, in the presence of hypoglycaemia, is the most useful test in identifying the cause of hypoglycaemia (see Fig. 17.2
Hypoglycaemia
GLUCOSE HOMOEOSTASIS IN THE FED AND THE POSTABSORPTIVE STATES
HYPOGLYCAEMIA
The neuroendocrine response to hypoglycaemia
Symptoms of hypoglycaemia
Acute neuroglycopenia (neurogenic)
Subacute neuroglycopenia
Chronic neuroglycopenia
CLASSIFICATION OF HYPOGLYCAEMIC DISORDERS
PRACTICAL APPROACH TO THE INVESTIGATION OF HYPOGLYCAEMIA
Evaluation of hypoglycaemia in persons without diabetes mellitus
Investigation of hypoglycaemia
Demonstration of hypoglycaemia
Measurement of blood glucose during spontaneous symptoms
Provocation tests
Prolonged fast test
Glucagon stimulation test
Mixed meal test
Identification of the cause of hypoglycaemia
Plasma insulin, C-peptide and proinsulin
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


