45
Cancer Chemotherapy and Cytotoxic Agents
This chapter will be most useful after having a basic understanding of the material in Chapter 60 General Principles of Cancer Chemotherapy and Chapter 61 Cytotoxic Agents in Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th Edition. In addition to the material presented here, Chapters 60 and 61 of the 12th Edition contain:
• A description of the cell cycle, and cell cycle checkpoints and regulation
• The clinical pharmacology and toxicities of specific agents
• The molecular structures of cytotoxic agents used to treat cancer and other diseases such as immune disorders
DRUGS INCLUDED IN THIS CHAPTER
• 2-Mercaptoethanesulfonate (mesna; MESNEX)—reacts with acrolein in urine and protects against severe hemorrhagic cystitis in high-dose cyclophosphamide regimens
• 5-Fluorouracil (5-FU)
• 6-Mercaptopurine (6-MP; PURINETHOL, others)
• 6-Thioguanine (6-TG)
• Altretamine (hexamethylmelamine; HEXALEN)
• Amifostine (WR-2721; ETHYOL, others)—thiophosphate cytoprotective agent (see cisplatin clinical toxicities)
• Arsenic trioxide (ATO; TRISENOX)
• Azacytidine (5-azacytidine)
• Bendamustine (TREANDA, others)
• Bleomycin (BLENOXANE, others)
• Busulfan (MYLERAN, BUSULFEX)
• Carmustine (BCNU; BICNU, GLIADEL)
• Capecitabine (XELODA)
• Carboplatin (CBDCA, JM-8; PARAPLATIN)
• Carboxypeptidase G2 (a methotrexate-cleaving enzyme used to reduce methotrexate in plasma)
• Chlorambucil (LEUKERAN)
• Cisplatin (PLATINOL, others)
• Cladribine (2-chlorodeoxyadenosine, 2-CdA; LEUSTATIN, others)
• Clofarabine (2-chloro-2′-fluoro-arabinosyladenine)
• Cyclophosphamide (LYOPHILIZED CYTOXAN, others)
• Cytarabine (1-β-D-arabinofuranosylcytosine, cytosine arabinoside, Ara-C; CYTOSAR-U, TARABINE PFS, DEPOCYT, others)
• Dacarbazine (DTIC; DTIC-DOME)
• Dactinomycin (actinomycin D; COSMEGEN)
• Daunorubicin (daunomycin, rubidomycin; CERUBIDINE, others)
• Decitabine (2′-deoxy-5-azacytidine)
• Docetaxel (TAXOTERE)
• Doxorubicin (ADRIAMYCIN)
• Epirubicin (ELLENCE, others)
• Estramustine (EMCYT)
• Etoposide (VP-16-213; VEPESID, others)
• Floxuridine (FUdR, fluorodeoxyuridine; FUDR, others)
• Fludarabine phosphate (FLUDARA, OFORTA)
• Gemcitabine (2′,2′-difluorodeoxycytidine; dFdC, GEMZAR)
• High-dose methotrexate with leucovorin rescue (HDM-L)
• Hydroxyurea (HU; HYDREA, DROXIA, others)
• Idarubicin (IDAMYCIN PFS)
• Ifosfamide (IFEX, others)
• Irinotecan (CPT-11; CAMPTOSAR, others)
• Ixabepilone (IXEMPRA)
• L-asparaginase (L-ASP; ELSPAR)
• Leucovorin (folinic acid, citrovorum factor, 5-formyltetrahydrofolate, N5-formyl FH4)
• Lometrexol
• Lomustine (CCNU; CeeNU)
• Mechlorethamine HCl (MUSTARGEN)
• Melphalan (ALKERAN)
• Methotrexate (amethopterin; RHEUMATREX, TREXALL, others)
• Mitomycin (mitomycin-C; MUTAMYCIN, others)
• Mitotane (o,p’-DDD)
• Mitoxantrone (NOVANTRONE, others)
• Nab-paclitaxel (ABRAXANE)
• Nelarabine (6-methoxy-arabinosyl-guanine)
• Oxaliplatin (ELOXATIN)
• Paclitaxel (TAXOL, others)
• Pegaspargase (PEG-L-Asparaginase; ONCASPAR)
• Pemetrexed (MTA; ALIMTA)
• Pentostatin (2’-deoxycoformycin)
• Pralatrexate (FOLOTYN)
• Procarbazine (MATULANE)
• Raltitrexed (TOMUDEX)
• Romidepsin (depsipeptide, FK228; ISTODAX)
• Semustine (methyl-CCNU)
• Streptozocin (streptozotocin; ZANOSAR)
• Temozolomide (TEMODAR)
• Teniposide (VM-26; VUMON)
• Thiotepa (triethylenethiophosphoramide; THIOPLEX, others)
• Topotecan (HYCAMTIN)
• Trabectedin (YONDELIS)—not currently approved in the United States
• Tretinoin (all-trans retinoic acid; ATRA)
• Trimetrexate (NEUTREXIN)
• Valrubicin (VALSTAR)
• Vinblastine sulfate ((VELBAN, others)
• Vincristine sulfate (VINCASAR PFS, others)
• Vinorelbine (NAVELBINE, others)
• Vorinostat (suberoylanilide hydroxamic acid, SAHA; ZOLINZA)
MECHANISMS OF CYTOTOXICITY
• Alkylating agents interfere with DNA integrity and function, and primarily induce cell death in rapidly proliferating tissues.
• Lethality of DNA alkylation depends on the following:
▶ The recognition of the adduct by DNA repair systems.
▶ The creation of DNA strand breaks by repair enzymes.
▶ An intact apoptotic response.
• In nondividing cells, DNA damage activates a checkpoint that depends on the presence of a normal p53 gene.
▶ Cells thus blocked in the G1/S interface either repair DNA alkylation or undergo apoptosis.
▶ Malignant cells with mutant or absent p53 fail to suspend cell-cycle progression, do not undergo apoptosis, and exhibit resistance to alkylating drugs.
• Bifunctional agents, in which cytotoxic effects predominate, cause DNA strand cross-linking.
• Monofunctional methylating agents (procarbazine, temozolomide) have greater capacity for mutagenesis and carcinogenesis:
▶ Simple methylation may be bypassed by DNA polymerases, leading to mispairing reactions that permanently modify DNA sequence; these can be transmitted to subsequent generations leading to mutagenesis and carcinogenesis.
LEARNING OBJECTIVES
 Understand the mechanisms of action of cytotoxic antineoplastic agents on tumor cells (see Figure 45-1).
Understand the mechanisms of action of cytotoxic antineoplastic agents on tumor cells (see Figure 45-1).
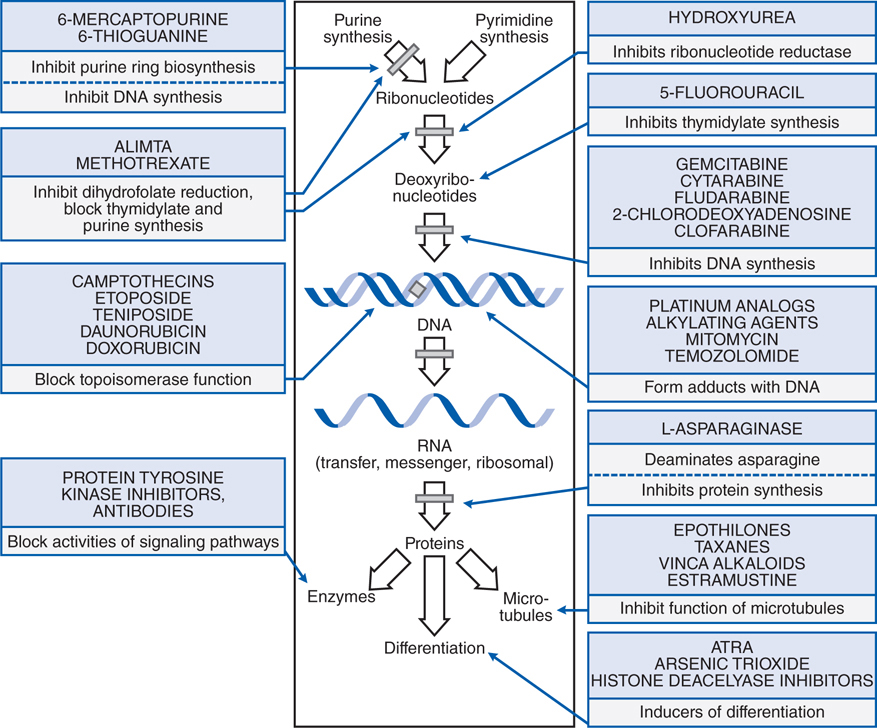
FIGURE 45-1 Summary of the mechanisms and sites of action of some chemotherapeutic agents useful in neoplastic disease.
 Understand the mechanisms of toxicity of cytotoxic antineoplastic agents on normal cells and strategies for reducing toxic effects.
Understand the mechanisms of toxicity of cytotoxic antineoplastic agents on normal cells and strategies for reducing toxic effects.
 Understand the mechanisms of drug resistance to individual agents and strategies to avoid resistance.
Understand the mechanisms of drug resistance to individual agents and strategies to avoid resistance.
 Know how pharmacogenetics can impact tumor sensitivity and toxicities of specific agents.
Know how pharmacogenetics can impact tumor sensitivity and toxicities of specific agents.
 Know the signs of acute toxicity and when chemotherapy should be altered or discontinued.
Know the signs of acute toxicity and when chemotherapy should be altered or discontinued.
 Know the risk of long-term toxicities that can occur with individual chemotherapeutic agents.
Know the risk of long-term toxicities that can occur with individual chemotherapeutic agents.
 Know the therapeutic strategies that can reduce acute and chronic toxicities.
Know the therapeutic strategies that can reduce acute and chronic toxicities.
 Know which classes of agents are typically used in treating specific cancers.
Know which classes of agents are typically used in treating specific cancers.
 Know which drugs are used in combination to improve tumor cell killing and reduce the risk of resistance development.
Know which drugs are used in combination to improve tumor cell killing and reduce the risk of resistance development.
A patient with a chronic lymphocytic leukemia is started on chemotherapy with cyclophosphamide.
a. What is the mechanism of action of cyclophosphamide?
Cyclophosphamide is a nitrogen mustard and is now the most widely used agent of this class of alkylating agents. Alkylating agents have in common the property of forming highly reactive carbonium ion intermediates. These reactive intermediates covalently link to sites of high electron density, such as phosphates, amines, sulfhydryl, and hydroxyl groups. Their chemotherapeutic and cytotoxic effects are directly related to the alkylation of reactive amine, oxygen, or phosphate groups on DNA (see Figure 45-1). The N7 atom of guanine is particularly susceptible to the formation of a covalent bond with bifunctional alkylating agents and may represent the key target that determines their biological effects (see Figure 45-2). Other atoms in the purine and pyrimidine bases of DNA, including N1 and N3 of the adenine ring, N3 of cytosine, and O6 of guanine, react with these agents, as do the amino and sulfhydryl groups of proteins and the sulfhydryl groups of glutathione.
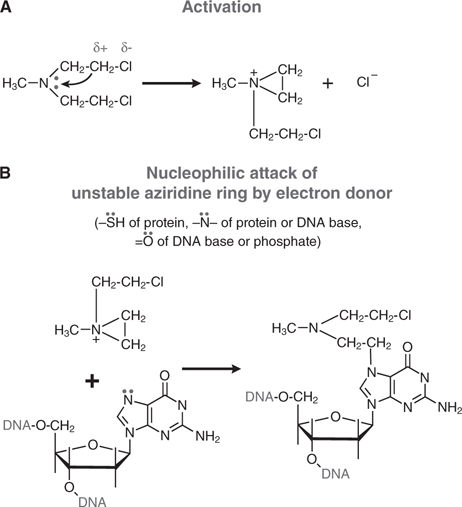
FIGURE 45-2 Mechanism of action of alkylating agents. A. Activation reaction. B. Alkylation of N7 of guanine.
MECHANISMS OF ACTION AND RESISTANCE OF ALKYLATING AGENTS AND PLATINUM COORDINATION COMPLEXES (SEE TABLE 45-1)
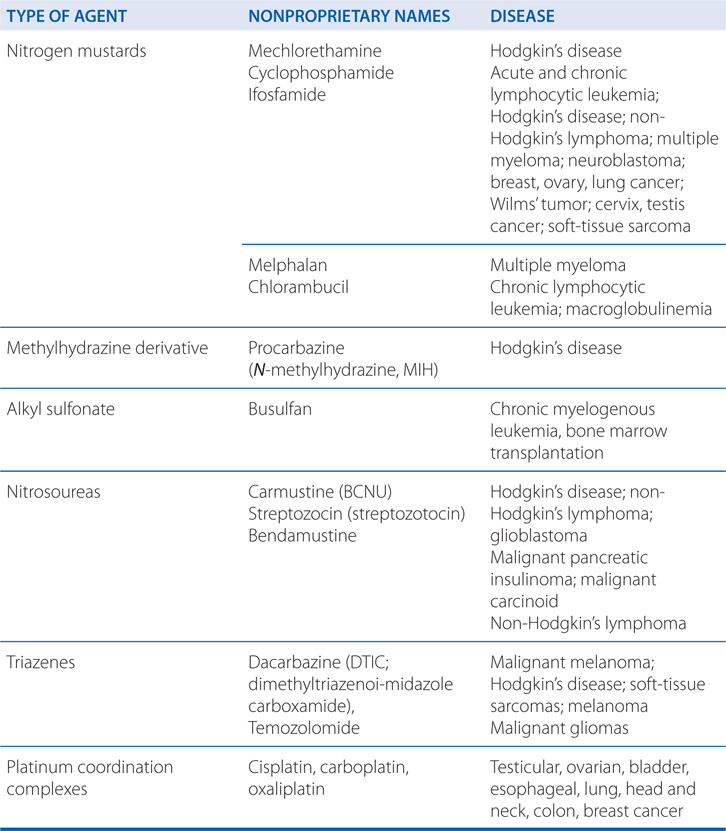
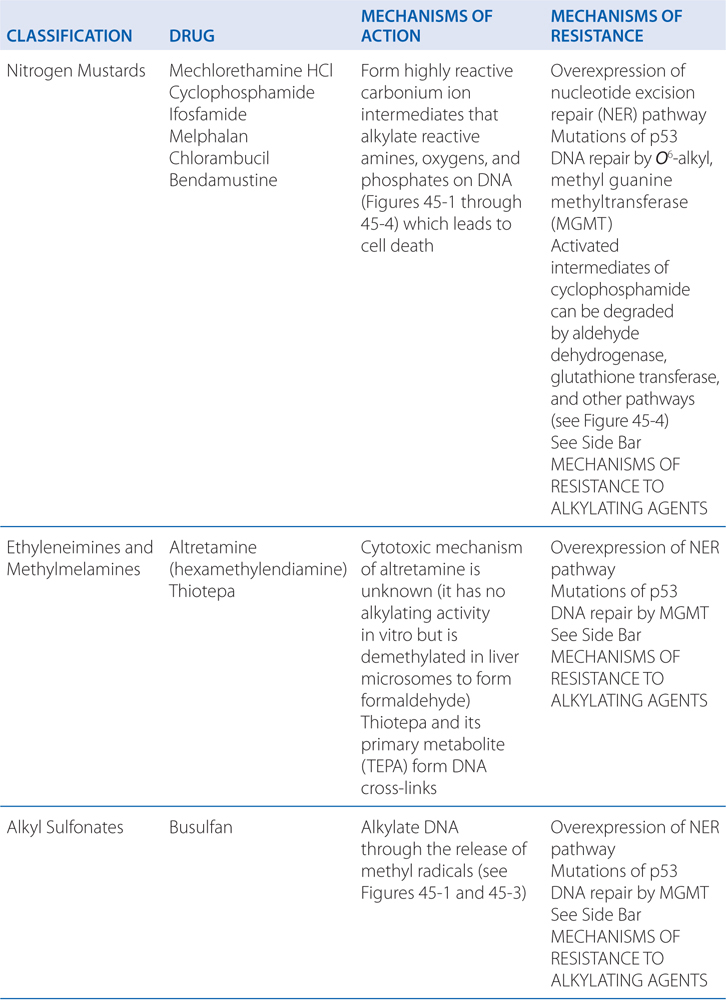
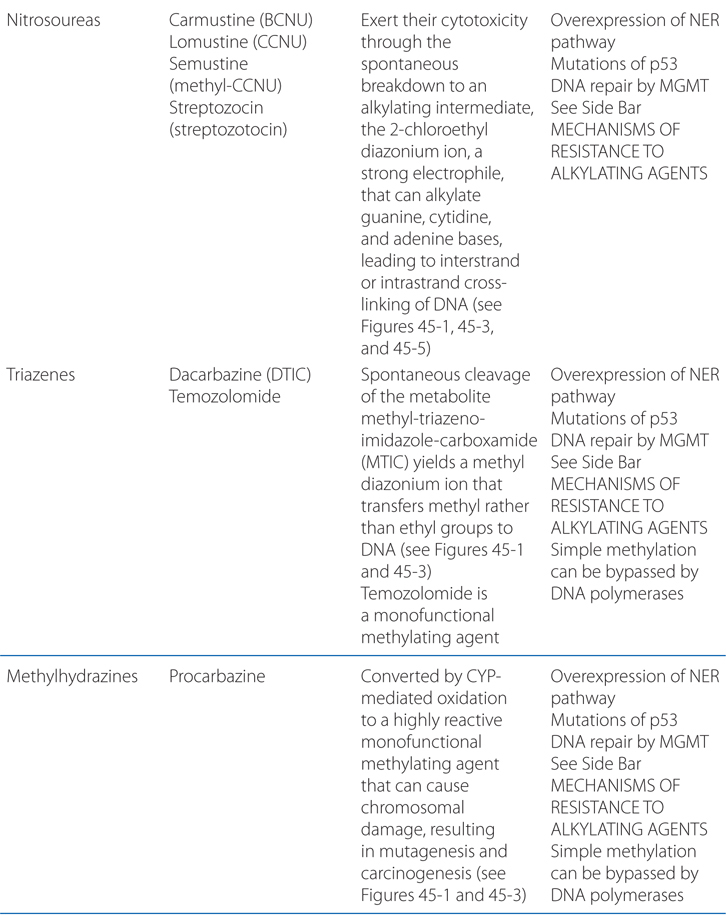
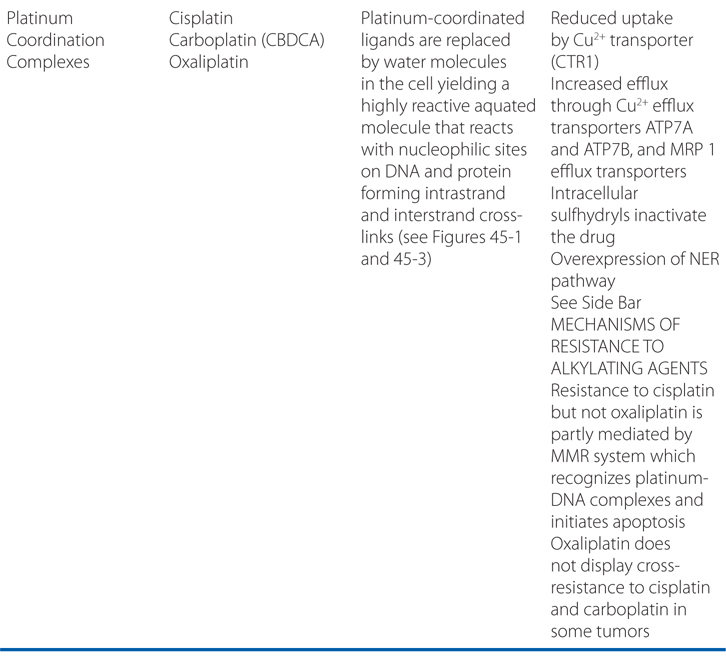
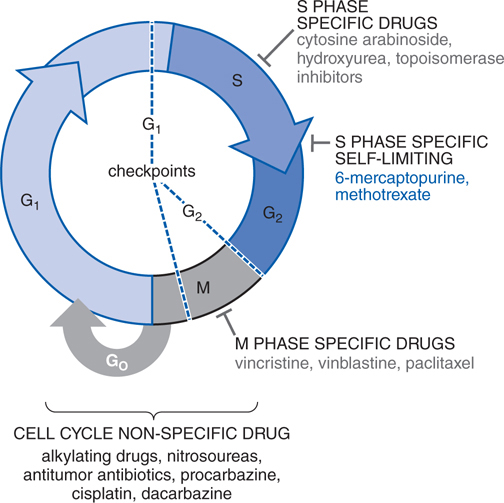
FIGURE 45-3 Cell cycle specificity of antineoplastic agents.
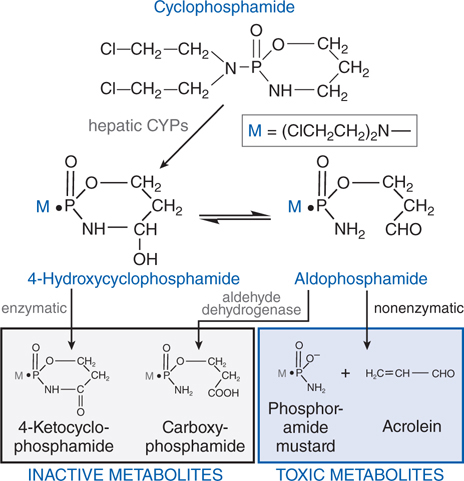
FIGURE 45-4 Metabolism of cyclophosphamide.
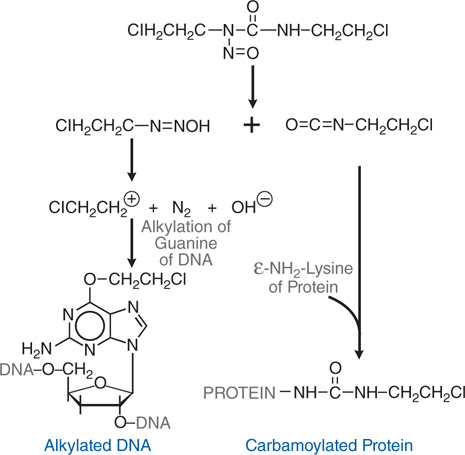
FIGURE 45-5 Degradation of carmustine (BCNU) with generation of alkylating and carbamylating intermediates.
Alkylation of the N7 of guanine in DNA exerts several biologically important effects. Guanine residues in DNA exist predominantly as the keto tautomer and readily make Watson-Crick base pairs by hydrogen bonding with cytosine residues. However, when the N7 of guanine is alkylated (to become a quaternary ammonium nitrogen), the guanine residue is more acidic and the enol tautomer is favored. The modified guanine can mispair with thymine residues during DNA synthesis, leading to the substitution of thymine for cytosine. Second, alkylation of the N7 creates lability in the imidazole ring, leading to opening of the ring and excision of the damaged guanine residue. Mispairing and imidazole ring opening can lead to attempts to repair the damaged stretch of DNA, causing strand breakage. Third, with bifunctional alkylating agents such as nitrogen mustards, the second 2-chloroethyl side chain can undergo a similar cyclization reaction and alkylate a second guanine residue or another nucleophilic moiety, resulting in the crosslinking of two nucleic acid chains or the linking of a nucleic acid to a protein, alterations that would cause a major disruption in nucleic acid function. Any of these effects could contribute to both the mutagenic and cytotoxic effects of alkylating agents. The extreme cytotoxicity of bifunctional alkylators correlates very closely with interstrand cross-linkage of DNA.
• Alkylation of a single strand of DNA (mono-adducts) is repaired by the nucleotide excision repair (NER) pathway.
• DNA strand cross-links require participation of nonhomologous end joining, an error-prone pathway, or the error-free homologous recombination pathway.
• The homologous recombination (end joining) pathway has multiple components:
▶ Sensors of DNA integrity (such as p53)
▶ Activation signals such as the ataxia-telangiectasia-mutated (ATM) and ataxia-telangiectasia and rad-related (ATR) proteins
▶ Activated DNA repair complex composed of Fanconi anemia proteins and BRCA2, all of which localize at the site of DNA damage and initiate removal of the cross-linked segment of DNA
▶ Homologous recombination, which allows re-synthesis of the damaged DNA sequence followed by religation of the repaired sequences
• The homologous recombination process depends on the presence and accurate functioning of multiple proteins; their absence or mutation, as in Fanconi anemia or ataxia telangiectasia, leads to extreme sensitivity to DNA cross-linking agents.
• Other repair enzymes are specific for removing methyl and ethyl adducts from the O6 of guanine (MGMT), and for repair of alkylation of the N-3 of adenine and N-7 of guanine (3-methyladenine-DNA glycosylase).
• For the methylating drugs, nitrosoureas, cisplatin and carboplatin, and thiopurine analogs, the mismatch repair (MMR) pathway is essential for cytotoxicity, causing strand breaks at sites of adduct formation, creating mispairing of thymine residues, and triggering apoptosis.
The capacity of alkylating agents to interfere with DNA integrity and function, and to induce cell death in rapidly proliferating tissues provides the basis for their therapeutic and toxic properties (see Side Bar MECHANISMS OF CYTOTOXICITY). Acute effects manifest primarily against rapidly proliferating tissues; however, certain alkylating agents may have damaging effects on tissues with normally low mitotic indices (eg, liver, kidney, and mature lymphocytes), which usually are affected in a delayed time frame.
b. How are the active alkylating species formed in vivo?
Cyclophosphamide is well absorbed orally and is activated to the 4-hydroxy intermediate in the liver by CYP2B (see Figure 45-4), with subsequent transport of the activated intermediate to sites of action. 4-Hydroxycyclophosphamide may be oxidized further by aldehyde oxidase, either in the liver or in tumor tissue, to inactive metabolites. 4-Hydroxycyclophosphamide and its tautomer, aldophosphamide, travel in the circulation to tumor cells where aldophosphamide cleaves spontaneously, generating stoichiometric amounts of phosphoramide mustard and acrolein. Phosphoramide mustard is responsible for antitumor effects, while acrolein is associated with significant toxicities.
c. What are the adverse effects of cyclophosphamide?
Alkylating agents are associated with a wide range of toxicities (see Side Bar TOXICITIES OF ALKYLATING DRUGS). Most alkylating agents (ie, melphalan, chlorambucil, cyclophosphamide, and ifosfamide) cause dose-limiting toxicity to bone marrow elements (myelosuppression) and, to a lesser extent, intestinal mucosa. Cyclophosphamide has lesser effects on peripheral blood platelet counts than do the other agents. Nausea and vomiting, which are due to neurotoxic effects, commonly follow agent administration of nitrogen mustard. High-dose therapy with cyclophosphamide can lead to organ damage, including GI ulceration, cystitis (see next paragraph), and, less commonly, pulmonary, renal, hepatic, and cardiac toxicities (a hemorrhagic myocardial necrosis). Dose-limiting major organ toxicities associated with alkylating agents are listed in Table 45-2.
TABLE 45-2 Dose-Limiting Extramedullary Toxicities of Single Alkylating Agents
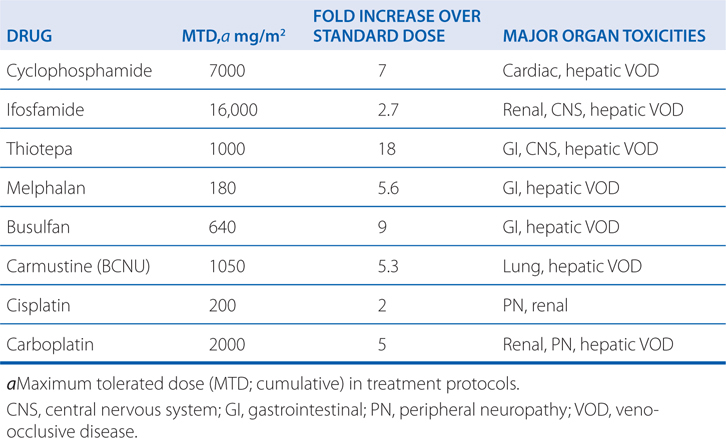
Cyclophosphamide and ifosfamide release a nephrotoxic and urotoxic metabolite, acrolein (see Figure 45-4), which causes a severe hemorrhagic cystitis in high-dose regimens. This adverse effect can be prevented by coadministration of 2-mercaptoethanesulfonate (mesna), which conjugates acrolein in urine. Mesna does not negate the systemic antitumor activity of the drug. Patients should receive vigorous intravenous hydration during high-dose treatment. Brisk hematuria in a patient receiving daily oral therapy should lead to immediate drug discontinuation. Refractory bladder hemorrhage can become life threatening, and cystectomy may be necessary for control of bleeding. Inappropriate secretion of antidiuretic hormone has been observed in patients receiving cyclophosphamide, usually at doses greater than 50 mg/kg. It is important to be aware of the possibility of water intoxication, because these patients usually are vigorously hydrated to prevent bladder toxicity.
Specific biochemical changes implicated in the development of resistance to an alkylating agent when used as a single agent include the following:
• Decreased permeation of actively transported drugs (mechlorethamine and melphalan).
• Increased intracellular concentrations of nucleophilic substances, principally thiols such as glutathione, which can conjugate with and detoxify electrophilic intermediates.
• Increased activity of DNA repair pathways, which may differ for the various alkylating agents:
▶ Increased activity of the complex NER pathway correlates with resistance to most chloroethyl and platinum adducts.
▶ MGMT activity determines response to BCNU and to methylating drugs such as the triazenes, procarbazine, temozolomide, and busulfan.
• Increased rates of metabolic degradation of the activated forms of cyclophosphamide and ifosfamide to their inactive keto and carboxy metabolites by aldehyde dehydrogenase (see Figure 45-4), and detoxification of most alkylating intermediates by glutathione transferases.
• Loss of the ability to recognize adducts formed by nitrosoureas and methylating agents, as the result of defective MMR capability, confers resistance, as does defective checkpoint function for virtually all alkylating drugs.
• Impaired apoptotic pathways (eg, overexpression of bcl-2) confer resistance.
All alkylating agents have toxic effects on the male and female reproductive systems, causing an often permanent amenorrhea, and an irreversible azoospermia in men.
Alkylating agents are highly leukemogenic. Acute nonlymphocytic leukemia peaks in incidence ~4 years after therapy and may affect up to 5% of patients treated on regimens containing alkylating drugs. Leukemia often is preceded by a period of neutropenia or anemia, and by bone marrow morphology consistent with myelodysplasia. Melphalan, the nitrosoureas, and the methylating agent procarbazine have the greatest propensity to cause leukemia, while it is less common after cyclophosphamide.
d. What are the mechanisms by which resistance to cyclophosphamide and other alkylating agents might develop?
The Side Bar MECHANISMS OF RESISTANCE TO ALKYLATING AGENTS describes mechanisms by which tumors can develop resistance to the alkylating agents. One mechanism of resistance is increased rates of metabolic degradation of the activated forms of cyclophosphamide to its inactive keto and carboxy metabolites by aldehyde dehydrogenase, as well as by inactivation by glutathione transferase and other enzymatic pathways (see Figure 45-4).
With all alkylating agents, lethality of DNA alkylation depends on the recognition of the DNA adduct, the creation of DNA strand breaks by repair enzymes, and an intact apoptotic response. The specific repair enzyme complex utilized will depend on two factors: the chemistry of the DNA adduct formed and the repair capacity of the cell involved. The process of recognizing and repairing DNA generally requires an intact NER complex. As described in the answer to Case 45-1a, the ability of the bifunctional nitrogen mustards to form interstrand DNA cross-links increases cytotoxicity over alkylation of single strands by monofunctional alkylating agents. Alkylation of a single strand of DNA (mono-adducts) is repaired by the NER pathway, while the less frequent DNA cross-links require participation of nonhomologous end joining, an error-prone pathway, or the error-free homologous recombination pathway (see Side Bar DNA REPAIR MECHANISMS).
Recognition of extensively damaged DNA by p53 can trigger apoptosis. In nondividing cells, DNA damage activates a checkpoint that depends on the presence of a normal p53 gene. Cells thus blocked in the G1/S interface either repair DNA alkylation or undergo apoptosis. Malignant cells with mutant or absent p53 fail to suspend cell-cycle progression, do not undergo apoptosis, and exhibit resistance to alkylating drugs.
TOXICITIES OF ALKYLATING DRUGS (See Table 45-2)
• Most alkylating agents cause dose-limiting toxicity to bone marrow elements, with acute myelosuppression that peaks at 6-10 days and recovers in 14-21 days.
• Alkylating agents are highly toxic to dividing mucosal cells and to hair follicles, leading to oral mucosal ulceration, intestinal denudation, and alopecia.
• Neurotoxic effects range from nausea and vomiting to more severe central and peripheral toxicities that include altered mental status, coma, generalized seizures, and cerebellar ataxia.
• Other organ toxicities may supervene after prolonged or high-dose use; these effects can appear after months or years and may be irreversible and even lethal:
▶ Pulmonary fibrosis.
▶ Vascular endothelial damage may precipitate veno-occlusive disease (VOD) of the liver, an often fatal side effect.
▶ Nephrotoxic and urotoxic (ie, toxic metabolites in the urine that can damage the bladder and lower urinary tract) effects.
▶ Unstable alkylating agents (particularly mechlorethamine and the nitrosoureas) have strong vesicant properties and cause damage to veins with repeated use and, if extravasated, produce ulceration.
▶ Toxic effects on the male and female reproductive systems, causing an often permanent amenorrhea, particularly in perimenopausal women, and an irreversible azoospermia in men.
• Alkylating agents are highly leukemogenic.
▶ Acute nonlymphocytic leukemia, often associated with partial or total deletions of chromosome 5 or 7, peaks in incidence ~4 years after therapy and may affect up to 5% of patients treated on regimens containing alkylating drugs.
Carboplatin, cisplatin, and oxaliplatin are referred to as platinum coordination complexes, and are widely used cancer chemotherapy agents.
a. What is the mechanism of action of these agents?
The platinum coordination complexes have broad antineoplastic activity and have become the foundation for the treatment of many cancers (see Table 45-1). They covalently bind to nucleophilic sites on DNA and share many pharmacological attributes with alkylators (see Figure 45-1).
Cisplatin, carboplatin, and oxaliplatin enter cells by an active Cu2+ transporter, CTR1. Inside the cell, the chloride, cyclohexane, or oxalate ligands of the three analogs are displaced by water molecules, yielding a positively charged and highly reactive molecule. The aquated species of the drug can react with electron-rich molecules, such as sulfhydryls, and with various sites on DNA, forming both intrastrand and interstrand cross-links. The N-7 of guanine is a particularly reactive site, leading to platinum cross-links between adjacent guanines (GG intrastrand cross-links) on the same DNA strand; guanine–adenine cross-links also form and may contribute to cytotoxicity. Interstrand cross-links form less frequently. DNA-platinum adducts inhibit replication and transcription, lead to single- and double-stranded breaks and miscoding, and, if recognized by p53 and other checkpoint proteins, cause induction of apoptosis. For cisplatin and carboplatin, the mismatch repair (MMR) pathway is essential for cytotoxicity, causing strand breaks at sites of adduct formation, creating mispairing of thymine residues, and triggering apoptosis.
The analogs differ in the conformation of their adducts and the effects of adduct on DNA structure and function. The oxaliplatin adducts are bulkier and less readily repaired, create a different pattern of distortion of the DNA helix, and differ from cisplatin adducts in the pattern of hydrogen bonding to adjacent segments of DNA. Unlike the other platinum analogs, oxaliplatin exhibits a cytotoxicity that does not depend on an active MMR system, which may explain its greater activity in colorectal cancer. It also seems less dependent on the presence of high-mobility group (HMG) proteins that are required by the other platinum derivatives.
b. How does the mechanism of action of these agents differ from the alkylating agents?
Cisplatin and other platinum complexes do not form carbonium ion intermediates like other alkylating agents or formally alkylate DNA; however, they covalently bind to nucleophilic sites on DNA causing DNA strand cross-links, and thus share many pharmacological attributes with alkylators.
c. What are the common toxicities of these agents?
Many of the toxicities of these agents are similar to the alkylating agents (see Side Bar TOXICITIES OF ALKYLATING DRUGS; also see Table 45-2). The primary clinical toxicities differ somewhat among cisplatin, carboplatin, and oxaliplatin.
An important dose-limiting toxicity of cisplatin is renal toxicity. To prevent renal toxicity, it is important to establish a chloride diuresis by the infusion of 1-2 L of normal saline prior to treatment. The appropriate amount of cisplatin then is diluted in a solution containing dextrose, saline, and mannitol, and administered over 4-6 hours. Amifostine is a thiophosphate cytoprotective agent that reduces renal toxicity associated with repeated administration of cisplatin, but is not commonly used.
Ototoxicity caused by cisplatin is unaffected by diuresis and is manifested by tinnitus and high-frequency hearing loss. The ototoxicity can be unilateral or bilateral, tends to be more frequent and severe with repeated doses, and may be more pronounced in children.
Marked nausea and vomiting occur in almost all patients receiving cisplatin and usually can be controlled with 5-HT3 antagonists, NK1-receptor antagonists, and high-dose corticosteroids (see Chapter 33). At higher doses, or after multiple cycles of treatment, cisplatin causes a progressive peripheral motor and sensory neuropathy, which may worsen after discontinuation of the drug and may be aggravated by subsequent or simultaneous treatment with taxanes or other neurotoxic drugs. Cisplatin causes mild to moderate myelosuppression, with transient leukopenia and thrombocytopenia. Anemia may become prominent after multiple cycles of treatment. Electrolyte disturbances, including hypomagnesemia, hypocalcemia, hypokalemia, and hypophosphatemia, are common. Hypocalcemia and hypomagnesemia secondary to tubular damage and renal electrolyte wasting may produce tetany if untreated. Routine measurement of Mg2+ concentrations in plasma is recommended. Hyperuricemia, hemolytic anemia, and cardiac abnormalities are rare side effects. Anaphylactic-like reactions, characterized by facial edema, bronchoconstriction, tachycardia, and hypotension, may occur within minutes after administration and should be treated by intravenous injection of epinephrine and with corticosteroids or antihistamines.
Carboplatin is relatively well tolerated clinically, causing less nausea, neurotoxicity, ototoxicity, and nephrotoxicity than cisplatin. Instead, the dose-limiting toxicity is myelosuppression, primarily thrombocytopenia. It is more likely to cause a hypersensitivity reaction; in patients with a mild reaction, premedication, graded doses of drug, and more prolonged infusion lead to desensitization. Carboplatin is an effective alternative for responsive tumors in patients unable to tolerate cisplatin because of impaired renal function, refractory nausea, significant hearing impairment, or neuropathy, but doses must be adjusted for renal function.
The dose-limiting toxicity of oxaliplatin is a peripheral neuropathy. An acute form, often triggered by exposure to cold liquids, manifests as paresthesias and/or dysesthesias in the upper and lower extremities, mouth, and throat. It may be caused by rapid release of oxalate, with depletion of calcium and magnesium, and responds to infusion of these electrolytes. A second type of peripheral neuropathy is more closely related to cumulative dose and has features similar to cisplatin neuropathy: progressive sensory neurotoxicity, with dysesthesias, ataxia, and numbness of the extremities. Hematological toxicity is mild to moderate, except for rare immune-mediated cytopenias, and nausea is well controlled with 5-HT3 receptor antagonists. Oxaliplatin may cause an acute allergic response with urticaria, hypotension, and bronchoconstriction.
All of the platinum analogs are mutagenic, teratogenic, and carcinogenic. Like other DNA adduct–forming drugs, these agents have been associated with pulmonary fibrosis and the development of leukemia, months to years after treatment.
d. What are the mechanisms for resistance and sensitivity to the platinum coordination complex agents?
Resistance to the platinum analogs likely is multifactorial (see Side Bar MECHANISMS OF RESISTANCE TO ALKYLATING AGENTS), and the compounds differ in their degree of cross-resistance. Carboplatin shares cross-resistance with cisplatin in most experimental tumors, while oxaliplatin does not.
Repair of platinum-DNA adducts requires participation of the NER pathway. Inhibition or loss of NER increases sensitivity to cisplatin in ovarian cancer patients, while overexpression of NER components is associated with poor response to cisplatin or oxaliplatin-based therapy in lung, colon, and gastric cancer. Higher levels of expression of the NER component ERCC1, in tumor cells and peripheral white blood cells are associated with a lower response rate in patients with solid tumors.
Resistance to cisplatin, but not oxaliplatin, appears to be partly mediated through loss of function in the MMR proteins (hMLH1, hMLH2, or hMSH6), which recognize platinum-DNA adducts and initiate apoptosis.
In the absence of effective repair of DNA-platinum adducts, sensitive cells cannot replicate or transcribe affected portions of the DNA strand. However, it is clear that some DNA polymerases can bypass adducts, especially those created by cisplatin. Oxaliplatin adducts are less easily bypassed. It remains unproven whether these polymerases contribute to resistance.
Platinum coordination complex drugs enter cells by active transport and cisplatin resistance related to loss of active uptake has been demonstrated in vitro. The compounds are actively extruded from cells by ATP7A and ATP7B copper transporters and by multidrug resistance protein 1 (MRP1/ABCC1); variable expression of these transporters may contribute to clinical resistance. Overexpression of copper efflux transporters, ATP7A and ATP7B, correlates with poor survival after cisplatin-based therapy for ovarian cancer.
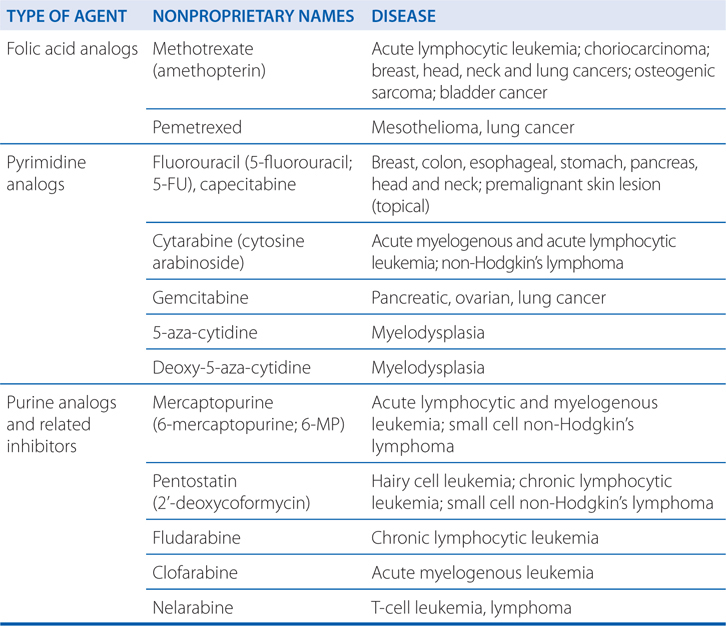
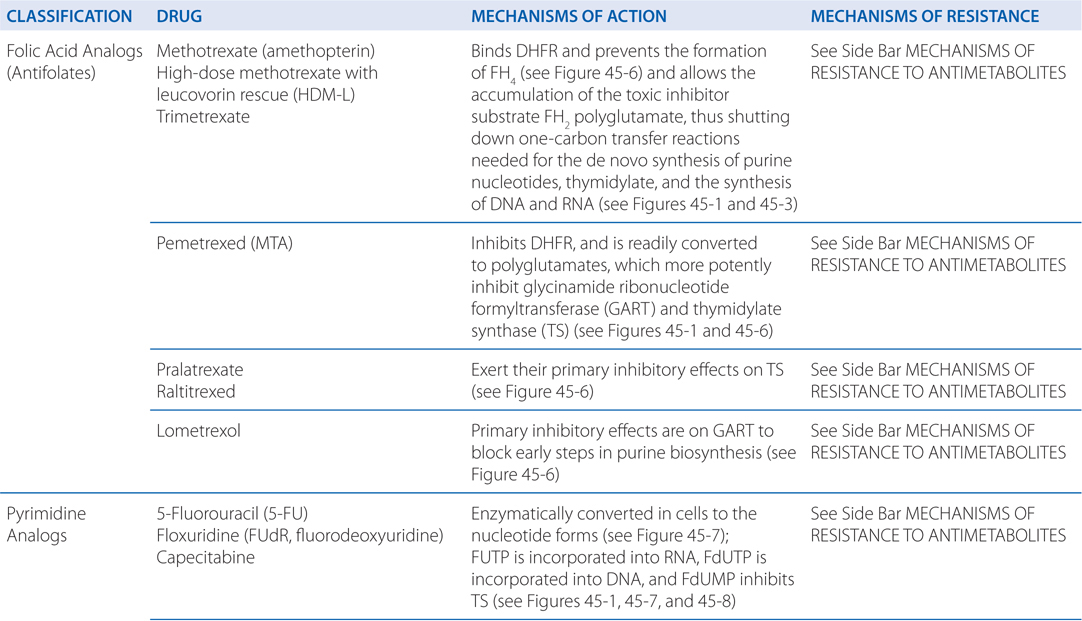
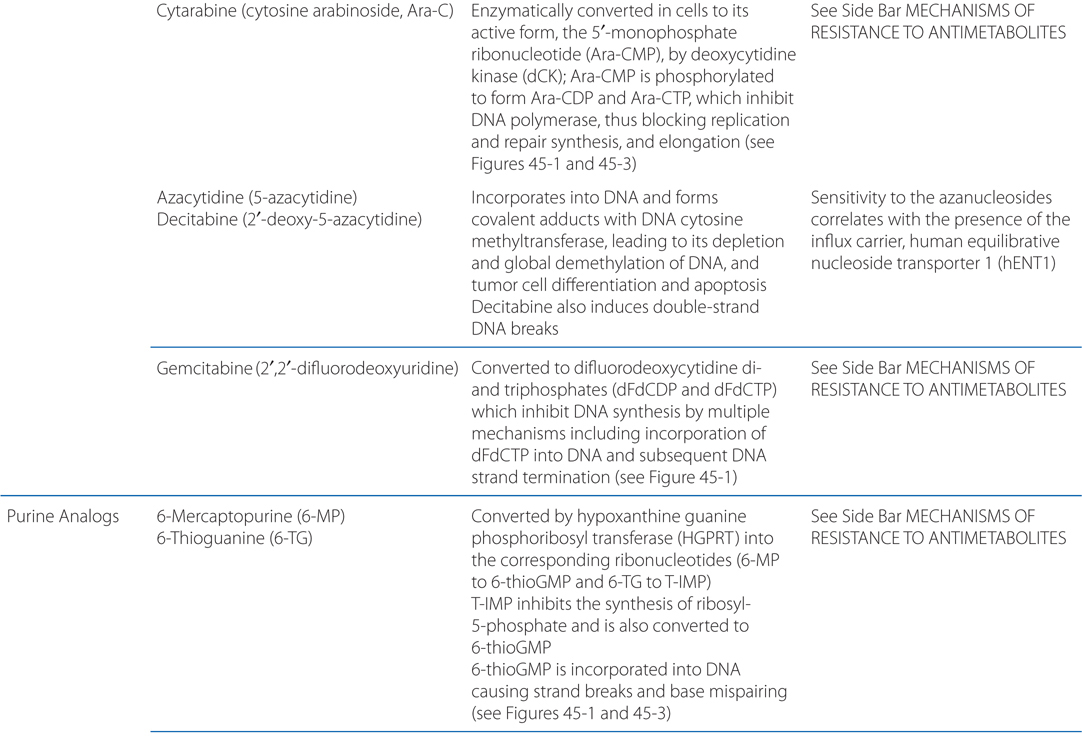
MECHANISMS OF ACTION AND RESISTANCE OF ANTIMETABOLITES (SEE TABLE 45-3)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


