TABLE 9.1 Osteoma: Clinical and Roentgenographic Types | |||||||
|---|---|---|---|---|---|---|---|
|
(b) osteomas, and (c) epidermal inclusion cysts of the skin. Osteomas in Gardner syndrome may or may not be well defined and has been reported in long bones, skull, mandible, and paranasal sinuses. Defective dentition and fibrous tumors may also be present. Polyposis in Gardner syndrome begins late in childhood or early in adulthood, and often results in gastrointestinal carcinoma.
 FIGURE 9.1. Exophthalmos of the right orbit (A) because of a large, well-circumscribed, ivory-white osteoma of the frontal sinus (B). |
 FIGURE 9.2. Skeletal distribution of parosteal osteoma. (From Bertoni F, Unni KK, Beabout JW, et al. Parosteal osteoma of bones other than of the skull and face. Cancer. 1995;75:2466-2473. Copyright © 1995, American Cancer Society. Reprinted with permission of Wiley-Liss, Inc.) |
(c) spongiosis—periphery of compact bone with radial septa and intervening marrow spaces; and (d) mixed—bone and fibrous tissue. The characterization into these subtypes is not clinically relevant.
 FIGURE 9.3. Osteoma. Gross and microscopic. An ivory-white osteoma appears nipple-like over the smooth calvarium of the skull (A); histologically, the lesion appears as a mature bone blending imperceptibly into the adjacent outer table of the skull (B); osteomas are usually mature cortical bone (C, left), but, while forming, will show foci of remodeling (C, right—open space). Osteoblastoma-like osteoma. Irregular spicules of bone in a fibrovascular stroma with active osteoblast and osteoclast remodeling. [(D) Roentgenograph; (E1 and E2) Microscopy]. |
 FIGURE 9.4. Skeletal distribution of osteoid osteoma. |
within the nidus (11). The relief of pain by aspirin is thought to be due to the prostaglandin-inhibiting action of aspirin, prostaglandins having been linked to lesional tissue in osteoid osteoma (12). Prostaglandins E2, F, and alpha have been reported in much larger-than-expected quantities by Makley and Dunn (12). Their link to pain may be either through vasodilatory effects with local blood vessel proliferation causing a pressure-type effect or through an effect on the bradykinin system (13). Prostaglandins may in fact produce or contribute to the production of osteoid osteoma, prostaglandin receptors having been found in bone cells. The remodeling bone effects of prostaglandins are well known and witnessed in other conditions such as mastocytosis (see Chapter 4).
 FIGURE 9.5. Osteoid osteoma. Roentgenographic appearance. (A) The left distal tibia shows distinct cortical sclerosis. (B) Tomographic films demonstrate a radiolucent nidus. (C) The lesion on CT scan shows cortical sclerosis and central nidus, within which is a radiodense (bone-forming) central core. (Continued) |
technetium injection can localize the lesion intraoperatively. Coupled with intraoperative localization by scintillation probe, conservative surgical excision can be accomplished and is of considerable benefit to the patient (Table 9.2).
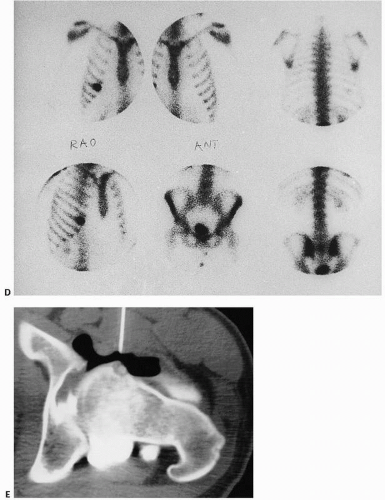 FIGURE 9.5. (Continued) (D) Lesions are very hot (black) on bone scan (rib). (E) CT scan. After double-contrast arthrography of the hip (the needle for the arthrogram is still in place), the nidus is seen in the anterior surface of the femoral neck. Small collection of calcium (bone) is present in the center of the lucent nidus. The lack of surrounding sclerosis is due to the intra-articular location of the lesion. |
Image-guided cryotherapy (19)
Interstitial laser photocoagulation (20)
CT-guided drilling with ethanol injection
CT-guided radiofrequency ablation (21)
Magnetic resonance-guided focused ultrasound (MRgFUS) (22).
 FIGURE 9.6. Osteoid osteoma (gross). Lesions consist of a well-defined focus of bone (nidus) surrounded by a reddish perimeter of less dense bone, all enveloped by dense sclerotic host bone (A, B). Large lesions have a gritty, cherry-red appearance (C). |
 FIGURE 9.7. Osteoid osteoma. The radiolucent nidus consists of a sclerotic dense central core within a less dense perimeter of bone. The nidus is surrounded by very sclerotic host bone. |
 FIGURE 9.8. Osteoid osteoma (microscopy). Identified by its small size and circumscription, osteoid osteoma is often buried within thickened cortical bone (A). The nidus (B, left) is distinct from the surrounding remodeling bone (B, right). (Continued) |
 FIGURE 9.8. (Continued) Osteoid osteoma nidus. The nidus is characterized by thinner sinewy streams of interlacing, often incompletely mineralized, trabecular bone rimmed by abundant osteoid and osteoblasts (C, D). Osteoblasts are often irregularly shaped. Osteoclasts are also seen. Intervening stroma is sparsely cellular and strikingly vascular, often fibrovascular in nature. |
 FIGURE 9.9. Osteoid osteoma. Experience in a series of operations attempting to localize the lesion. (Modified after Sim FH, Dahlin DC, Beabout JW. Osteoid-osteoma: diagnostic problems. J Bone Joint Surg Am. 1975;57:154-159.) |
 FIGURE 9.10. Osteoid osteoma. Minute lesion measuring just a few millimeters identified by serially sectioning and x-raying the removed specimens. |
TABLE 9.2 Results of Procedures Used to Localize and Diagnose Osteoid Osteomaa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 FIGURE 9.11. Osteoid osteoma versus osteoblastoma versus osteogenic sarcoma. |
osteoblasts, with deeply staining eosinophilic cytoplasm found in aggregates or sheet-like proliferation. Nuclei may be quite large, with vesicular finely clumped chromatin. Stromal mitoses are noted as well as a disorganized pattern of osteoid and osteoclasts. Cartilage and calcified cartilage are not usually encountered. In differentiating osteoblastoma from osteosarcoma, osteoblastoma stroma always has a benign fibrovascular appearance in the intervening spaces between bone production.
 FIGURE 9.12. Osteoblastoma (microscopy). Interlacing spicules of osteoid and irregular and mineralized cellular bone with abundant osteoblasts (A) and intervening hypervascular stroma (B). More aggressive osteoblastomas are characterized by “epithelioid” osteoblasts, plump epithelial-like osteoblasts (C). In osteoblastoma-like osteosarcoma, lesions are less well circumscribed (D) and, on high-power microscopy, reveal more nuclear pleomorphism than in osteoblastomas (E). |
 FIGURE 9.13. Skeletal distribution of osteoblastoma. |
 FIGURE 9.14. Osteoblastoma. Roentgenographic appearance. Radiodense destructive mass destroying the pedicle and transverse process of the right side of the lumbar fourth vertebra (A). Poorly defined sclerotic mass of the proximal ulna (B). Mildly expansile partially calcified lesion in the lamina of a lumbar vertebrae (mimicking a “giant osteoid osteoma”) (C). |
TABLE 9.3 Clinical Differentiation: Osteoid Osteoma and Osteoblastoma (Based on 860 Osteoid Osteomas and 184 Osteoblastomas) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
TABLE 9.4 Aggressive Osteoblastoma versus Osteosarcoma | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
 FIGURE 9.15. Skeletal distribution of osteosarcoma. |
a malignant pathologic fracture (SUVmax 12.0, range 4 to 45 for malignant) versus benign (SUVmax 2.9, range 0.6 to 5.5) has been used (43). For distinguishing benign tumors from malignant ones, a SUVmax of 6.8 ± 4.7 for malignant tumors and 4.5 ± 3.3 for benign lesions have been used, notably not statistically significant findings (44). PET scanning has also been used to evaluate tumor necrosis after chemotherapy with high SUVmax values in one study correlated with poor survival (45).
 FIGURE 9.16. Osteosarcoma. Roentgenographic appearance. Lesions of the distal femur (A), proximal humerus (B), and proximal femur (C). In the distal femur (A), a large poorly defined mixed lucent and sclerotic lesion extends from the metaphysis to the diaphysis. Epiphysis is spared. Tumor has broken through the lateral surface. Extensive periosteal new bone is seen both metaphyseal and diaphyseal. Because they are bone forming, osteosarcomas are hot on a bone scan (D). |
metastases has improved the outlook considerably in children with osteosarcoma and synchronous pulmonary metastases. Since 1982, there has been a 50 percent probability of survival at 3 years, compared with no survival prior to that time (63).
 FIGURE 9.17. Osteosarcoma (gross). Distal femur (A), proximal humerus (B), and distal femur (C) with sclerotic lesions of the metaphysis destroying cortical bone and extending into soft tissue. Growth plate is a relative barrier. Color varies from ivory-white to yellow to red, depending on the amount of bone, necrosis, and hemorrhage. |
With the advent of neoadjuvant chemotherapy and surgical removal, the 5- and 10-year survivals of children with osteogenic sarcoma without evidence of disseminated disease at diagnosis is considerably higher and is in the 70 percent to 80 percent range.
doxorubicin,
cisplatin, and
high-dose metotrexate.
 FIGURE 9.18. Osteosarcoma (gross). Codman triangle. Distal femur with tumor raising the periosteum. (A) Low power and specimen x-ray. (B) Higher power. Borders of Codman triangle are the bone surface, the tumor, and the periosteal surface. |
 FIGURE 9.19. Osteosarcoma (histopathology). Bone marrow is totally replaced by tumor (A), which, at high power, reveals pleomorphic, mitotic cells making malignant osteoid (B). Osteosarcomas demonstrate a wide variety of histologic features, with fibrous and chondroid areas suggesting a painter’s palette (C, D). (Continued)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|