KEY POINTS
Wound healing is a complex cellular and biochemical cascade that leads to restitution of integrity and function.
Although individual tissues may have unique healing characteristics, all tissues heal by similar mechanisms, and the process undergoes phases of inflammation, cellular migration, proliferation, matrix deposition, and remodeling.
Factors that impede normal healing include local, systemic, and technical conditions that the surgeon must take into account.
Clinically, excess healing can be as significant a problem as impaired healing; genetic, technical, and local factors play a major role.
Optimal outcome of acute wounds relies on complete evaluation of the patient and of the wound and application of best practices and techniques.
HISTORY OF WOUND HEALING
The earliest accounts of wound healing date back to about 2000 b.c., when the Sumerians employed two modes of treatment: a spiritual method consisting of incantations, and a physical method of applying poultice-like materials to the wound. The Egyptians were the first to differentiate between infected and diseased wounds compared to noninfected wounds. The 1650 b.c. Edwin Smith Surgical Papyrus, a copy of a much older document, describes at least 48 different types of wounds. A later document (Ebers Papyrus, 1550 b.c.) relates the use of concoctions containing honey (antibacterial properties), lint (absorbent properties), and grease (barrier) for treating wounds. These same properties are still considered essential in contemporary daily wound management.
The Greeks, equipped with the knowledge bequeathed by the Egyptians, went even further and classified wounds as acute or chronic in nature. Galen of Pergamum (120–201 a.d.), appointed as the doctor to the Roman gladiators, had an enormous number of wounds to deal with following gladiatorial combats. He emphasized the importance of maintaining a moist environment to ensure adequate healing. It took almost 19 centuries for this important concept to be proven scientifically, when it was shown that the epithelialization rate increases by 50% in a moist wound environment when compared to a dry wound environment.1
The next major stride in the history of wound healing was the discovery of antiseptics and their importance in reducing wound infections. Ignaz Philipp Semmelweis, a Hungarian obstetrician (1818–1865), noted that the incidence of puerperal fever was much lower if medical students, following cadaver-dissection class and prior to attending childbirth, washed their hands with soap and hypochlorite. Louis Pasteur (1822–1895) was instrumental in dispelling the theory of spontaneous generation of germs and proving that germs existed in and were always introduced from the environment. Joseph Lister probably made one of the most significant contributions to wound healing. On a visit to Glasgow, Scotland, Lister noted that some areas of the city’s sewer system were less murky than the rest. He discovered that the water from pipes that were dumping waste containing carbolic acid (phenol) was clear. In 1865, Lister began soaking his surgical instruments in phenol and spraying the operating rooms, reducing the postoperative mortality rates from 50% to 15%. After attending an impressive lecture by Lister in 1876, Robert Wood Johnson left the meeting and began 10 years of research that would ultimately result in the production of an antiseptic dressing in the form of cotton gauze impregnated with iodoform. Since then, several other materials have been used to impregnate cotton gauze to achieve antisepsis.
The 1960s and 1970s led to the development of polymeric dressings. These polymeric dressings can be custom made to specific parameters, such as permeability to gases (occlusive vs. semiocclusive), varying degrees of absorbency, and different physical forms. Due to the ability to customize, the available range of materials that aid in wound care has grown exponentially to include an ever-expanding variety. Currently, the practice of wound healing encompasses manipulation and/or use of, among others, inflammatory cytokines, growth factors, and bioengineered tissue. It is the combination of all these modalities that enables optimal wound healing.
PHASES OF WOUND HEALING
As noted by John Hunter (1728–1793), a keen observer of biologic phenomena, “… the injury alone has in all cases a tendency to produce the disposition and the means of a cure.”2 Normal wound healing follows a predictable pattern that can be divided into overlapping phases defined by characteristic cellular populations and biochemical activities: (a) hemostasis and inflammation, (b) proliferation, and (c) maturation and remodeling. An approximate timeline of these events is depicted in Fig. 9-1. This sequence of events is fluid and overlapping, and in most circumstances spans the time from injury to resolution of acute wounds. All wounds need to progress through this series of cellular and biochemical events that characterizes the phases of healing in order to successfully re-establish tissue integrity.
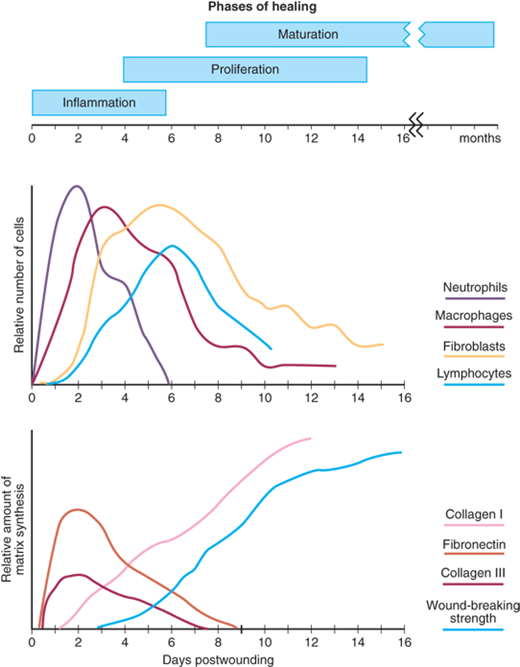
Hemostasis precedes and initiates inflammation with the ensuing release of chemotactic factors from the wound site (Fig. 9-2A). Wounding by definition disrupts tissue integrity, leading to division of blood vessels and direct exposure of extracellular matrix to platelets. Exposure of subendothelial collagen to platelets results in platelet aggregation, degranulation, and activation of the coagulation cascade. Platelet α granules release a number of wound-active substances, such as platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), platelet-activating factor (PAF), fibronectin, and serotonin. In addition to achieving hemostasis, the fibrin clot serves as scaffolding for the migration into the wound of inflammatory cells such as polymorphonuclear leukocytes (PMNs, neutrophils) and monocytes.

Cellular infiltration after injury follows a characteristic, predetermined sequence (see Fig. 9-1). PMNs are the first infiltrating cells to enter the wound site, peaking at 24 to 48 hours. Increased vascular permeability, local prostaglandin release, and the presence of chemotactic substances such as complement factors, interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), TGF-β, platelet factor 4, or bacterial products all stimulate neutrophil migration.
The postulated primary role of neutrophils is phagocytosis of bacteria and tissue debris. PMNs are also a major source of cytokines early during inflammation, especially TNF-α3 which may have a significant influence on subsequent angiogenesis and collagen synthesis (see Fig. 9-2B). PMNs also release proteases such as collagenases, which participate in matrix and ground substance degradation in the early phase of wound healing. Other than their role in limiting infections, these cells do not appear to play a role in collagen deposition or acquisition of mechanical wound strength. On the contrary, neutrophil factors have been implicated in delaying the epithelial closure of wounds.4
The second population of inflammatory cells that invades the wound consists of macrophages, which are recognized as being essential to successful healing.5 Derived from circulating monocytes, macrophages achieve significant numbers in the wound by 48 to 96 hours postinjury and remain present until wound healing is complete.
Macrophages, like neutrophils, participate in wound débridement via phagocytosis and contribute to microbial stasis via oxygen radical and nitric oxide synthesis (see Fig. 9-2B,C). The macrophage’s most pivotal function is activation and recruitment of other cells via mediators such as cytokines and growth factors, as well as directly by cell-cell interaction and intercellular adhesion molecules (ICAM). By releasing such mediators as TGF-β, vascular endothelial growth factor (VEGF), insulin-like growth factor (IGF), epithelial growth factor (EGF), and lactate, macrophages regulate cell proliferation, matrix synthesis, and angiogenesis.6,7 Macrophages also play a significant role in regulating angiogenesis and matrix deposition and remodeling (Table 9-1).
ACTIVITY | MEDIATORS |
|---|---|
Phagocytosis | Reactive oxygen species Nitric oxide |
Débridement | Collagenase, elastase |
Cell recruitment and activation | Growth factors: PDGF, TGF-β, EGF, IGF Cytokines: TNF-α, IL-1, IL-6 Fibronectin |
Matrix synthesis | Growth factors: TGF-β, EGF, PDGF Cytokines: TNF-α, IL-1, IFN-γ Enzymes: arginase, collagenase Prostaglandins Nitric oxide |
Angiogenesis | Growth factors: FGF, VEGF Cytokines: TNF-α Nitric oxide |
T lymphocytes comprise another population of inflammatory/immune cells that routinely invades the wound. Less numerous than macrophages, T-lymphocyte numbers peak at about 1 week postinjury and truly bridge the transition from the inflammatory to the proliferative phase of healing. Though known to be essential to wound healing, the role of lymphocytes in wound healing is not fully defined.8 A significant body of data supports the hypothesis that T lymphocytes play an active role in the modulation of the wound environment. Depletion of most wound T lymphocytes decreases wound strength and collagen content,9 while selective depletion of the CD8+ suppressor subset of T lymphocytes enhances wound healing. However, depletion of the CD4+ helper subset has no effect.10 Lymphocytes also exert a downregulating effect on fibroblast collagen synthesis by cell-associated interferon (IFN)-γ, TNF-α, and IL-1. This effect is lost if the cells are physically separated, suggesting that extracellular matrix synthesis is regulated not only via soluble factors but also by direct cell-cell contact between lymphocytes and fibroblasts.11
The proliferative phase is the second phase of wound healing and roughly spans days 4 through 12 (see Fig. 9-2C). It is during this phase that tissue continuity is re-established. Fibroblasts and endothelial cells are the last cell populations to infiltrate the healing wound, and the strongest chemotactic factor for fibroblasts is PDGF.12,13 Upon entering the wound environment, recruited fibroblasts first need to proliferate, and then become activated, to carry out their primary function of matrix synthesis remodeling. This activation is mediated mainly by the cytokines and growth factors released from wound macrophages.
Fibroblasts isolated from wounds synthesize more collagen than nonwound fibroblasts, they proliferate less, and they actively carry out matrix contraction. Although it is clear that the cytokine-rich wound environment plays a significant role in this phenotypic alteration and activation, the exact mediators are only partially characterized.14,15 Additionally, lactate, which accumulates in significant amounts in the wound environment over time (~10 mmol), is a potent regulator of collagen synthesis through a mechanism involving adenosine diphosphate (ADP)-ribosylation.16,17
Endothelial cells also proliferate extensively during this phase of healing. These cells participate in the formation of new capillaries (angiogenesis), a process essential to successful wound healing. Endothelial cells migrate from intact venules close to the wound. Their migration, replication, and new capillary tubule formation is under the influence of such cytokines and growth factors as TNF-α, TGF-β, and VEGF. Although many cells produce VEGF, macrophages represent a major source in the healing wound, and VEGF receptors are located specifically on endothelial cells.18,19
Collagen, the most abundant protein in the body, plays a critical role in the successful completion of adult wound healing. Its deposition, maturation, and subsequent remodeling are essential to the functional integrity of the wound.
Although there are at least 18 types of collagen described, the main ones of interest to wound repair are types I and III. Type I collagen is the major component of extracellular matrix in skin. Type III, which is also normally present in skin, becomes more prominent and important during the repair process.
Biochemically, each chain of collagen is composed of a glycine residue in every third position. The second position in the triplet is made up of proline or lysine during the translation process. The polypeptide chain that is translated from mRNA contains approximately 1000 amino acid residues and is called protocollagen. Release of protocollagen into the endoplasmic reticulum results in the hydroxylation of proline to hydroxyproline and of lysine to hydroxylysine by specific hydroxylases (Fig. 9-3). Prolyl hydroxylase requires oxygen and iron as cofactors, α-ketoglutarate as co-substrate, and ascorbic acid (vitamin C) as an electron donor. In the endoplasmic reticulum, the protocollagen chain is also glycosylated by the linking of galactose and glucose at specific hydroxylysine residues. These steps of hydroxylation and glycosylation alter the hydrogen bonding forces within the chain, imposing steric changes that force the protocollagen chain to assume an α-helical configuration. Three α-helical chains entwine to form a right-handed superhelical structure called procollagen. At both ends, this structure contains nonhelical peptide domains called registration peptides. Although initially joined by weak, ionic bonds, the procollagen molecule becomes much stronger by the covalent cross-linking of lysine residues.
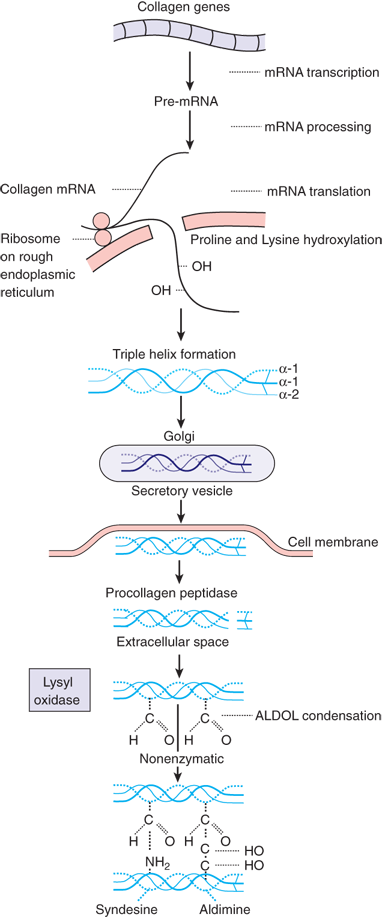
Extracellularly, the nonhelical registration peptides are cleaved by a procollagen peptidase, and the procollagen strands undergo further polymerization and cross-linking. The resulting collagen monomer is further polymerized and cross-linked by the formation of intra- and intermolecular covalent bonds.
Collagen synthesis, as well as posttranslational modifications, are highly dependent on systemic factors such as an adequate oxygen supply; the presence of sufficient nutrients (amino acids and carbohydrates) and cofactors (vitamins and trace metals); and the local wound environment (vascular supply and lack of infection). Addressing these factors and reversing nutritional deficiencies can optimize collagen synthesis and deposition.
Glycosaminoglycans comprise a large portion of the “ground substance” that makes up granulation tissue. Rarely found free, they couple with proteins to form proteoglycans. The polysaccharide chain is made up of repeating disaccharide units composed of glucuronic or iduronic acid and a hexosamine, which is usually sulfated. The disaccharide composition of proteoglycans varies from about 10 units in the case of heparan sulfate to as much as 2000 units in the case of hyaluronic acid.
The major glycosaminoglycans present in wounds are dermatan and chondroitin sulfate. Fibroblasts synthesize these compounds, increasing their concentration greatly during the first 3 weeks of healing. The interaction between collagen and proteoglycans is being actively studied. It is thought that the assembly of collagen subunits into fibrils and fibers is dependent upon the lattice provided by the sulfated proteoglycans. Furthermore, it appears that the extent of sulfation is critical in determining the configuration of the collagen fibrils. As scar collagen is deposited, the proteoglycans are incorporated into the collagen scaffolding. However, with scar maturation and collagen remodeling, the content of proteoglycans gradually diminishes.
The maturation and remodeling of the scar begins during the fibroplastic phase and is characterized by a reorganization of previously synthesized collagen. Collagen is broken down by matrix metalloproteinases (MMPs), and the net wound collagen content is the result of a balance between collagenolysis and collagen synthesis. There is a net shift toward collagen synthesis and eventually the re-establishment of extracellular matrix composed of a relatively acellular collagen-rich scar.
Wound strength and mechanical integrity in the fresh wound are determined by both the quantity and quality of the newly deposited collagen. The deposition of matrix at the wound site follows a characteristic pattern: fibronectin and collagen type III constitute the early matrix scaffolding; glycosaminoglycans and proteoglycans represent the next significant matrix components; and collagen type I is the final matrix. By several weeks postinjury, the amount of collagen in the wound reaches a plateau, but the tensile strength continues to increase for several more months.20 Fibril formation and fibril cross-linking result in decreased collagen solubility, increased strength, and increased resistance to enzymatic degradation of the collagen matrix. Fibrillin, a glycoprotein secreted by fibroblasts, is essential for the formation of elastic fibers found in connective tissue. Scar remodeling continues for many (6 to 12) months postinjury, gradually resulting in a mature, avascular, and acellular scar. The mechanical strength of the scar never achieves that of the uninjured tissue.
There is a constant turnover of collagen in the extracellular matrix, both in the healing wound as well as during normal tissue homeostasis. Collagenolysis is the result of collagenase activity, a class of MMPs that require activation. Both collagen synthesis and lysis are strictly controlled by cytokines and growth factors. Some factors affect both aspects of collagen remodeling. For example, TGF-β increases new collagen transcription and also decreases collagen breakdown by stimulating synthesis of tissue inhibitors of metalloproteinase.21 This balance of collagen deposition and degradation is the ultimate determinant of wound strength and integrity.
While tissue integrity and strength are being re-established, the external barrier must also be restored. This process is characterized primarily by proliferation and migration of epithelial cells adjacent to the wound (Fig. 9-4). The process begins within 1 day of injury and is seen as thickening of the epidermis at the wound edge. Marginal basal cells at the edge of the wound lose their firm attachment to the underlying dermis, enlarge, and begin to migrate across the surface of the provisional matrix. Fixed basal cells in a zone near the cut edge undergo a series of rapid mitotic divisions, and these cells appear to migrate by moving over one another in a leapfrog fashion until the defect is covered.22 Once the defect is bridged, the migrating epithelial cells lose their flattened appearance, become more columnar in shape, and increase their mitotic activity. Layering of the epithelium is re-established, and the surface layer eventually keratinizes.23
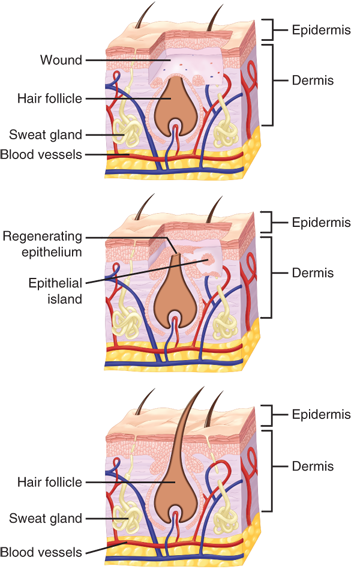
Re-epithelialization is complete in less than 48 hours in the case of approximated incised wounds, but may take substantially longer in the case of larger wounds, where there is a significant epidermal/dermal defect. If only the epithelium and superficial dermis are damaged, such as occurs in split-thickness skin graft donor sites or in superficial second-degree burns, then repair consists primarily of re-epithelialization with minimal or no fibroplasia and granulation tissue formation. The stimuli for re-epithelialization remain incompletely defined; however, it appears that the process is mediated by a combination of a loss of contact inhibition; exposure to constituents of the extracellular matrix, particularly fibronectin; and cytokines produced by immune mononuclear cells.24,25 In particular EGF, TGF-β, basic fibroblast growth factor (bFGF), PDGF, and IGF-1 have been shown to promote epithelialization.
Growth factors and cytokines are polypeptides produced in normal and wounded tissue that stimulate cellular migration, proliferation, and function. They often are named for the cells from which they were first derived (e.g., platelet-derived growth factor, PDGF) or for their initially identified function (e.g., fibroblast growth factor, FGF). These names are often misleading because growth factors have been demonstrated to have multiple functions. Most growth factors are extremely potent and produce significant effects in nanomolar concentrations.
They may act in an autocrine manner (where the growth factor acts on the cell producing it), a paracrine manner (by release into the extracellular environment, where it acts on the immediately neighboring cells), or in an endocrine manner (where the effect of the substance is distant to the site of release, and the substance is carried to the effector site through the blood stream). The timing of release may be as important as concentration in determining the effectiveness of growth factors. As these polypeptides exert their effects by cell-surface receptor binding, the appropriate receptor on the responding cells must be present at the time of release in order for the biologic effect to occur. Table 9-2 summarizes the principal growth factors found in healing wounds and their known effects on cells participating in the healing process. Growth factors have divergent actions on different cells; they can be chemoattractive to one cell type while stimulating replication of a different cell type. Little is known about the ratio of growth factor concentrations, which may be as important as the absolute concentration of individual growth factors.
GROWTH FACTOR | WOUND CELL ORIGIN | CELLULAR AND BIOLOGIC EFFECTS |
|---|---|---|
PDGF | Platelets, macrophages, monocytes, smooth muscle cells, endothelial cells | Chemotaxis: fibroblasts, smooth muscle, monocytes, neutrophils Mitogenesis: fibroblasts, smooth muscle cells Stimulation of angiogenesis Stimulation of collagen synthesis Enhance re-epithelization Modulate tissue remodeling |
FGF | Fibroblasts, endothelial cells, keratinocytes, smooth muscle cells, chondrocytes | Stimulation of angiogenesis (by stimulation of endothelial cell proliferation and migration) Mitogenesis: mesoderm and neuroectoderm |
HGF | Fibroblasts | Stimulates fibroblasts, keratinocytes, chondrocytes, myoblasts Suppresses inflammation, granulation tissue formation, angiogenesis, re-epithelialization |
Keratinocyte growth factor | Keratinocytes, fibroblasts | Significant homology with FGF; stimulates keratinocytes |
EGF | Platelets, macrophages, monocytes (also identified in salivary glands, duodenal glands, kidney, and lacrimal glands) | Stimulates proliferation and migration of all epithelial cell types |
TGF-α | Keratinocytes, platelets, macrophages | Homology with EGF; binds to EGF receptor Mitogenic and chemotactic for epidermal and endothelial cells |
TGF-β (three isoforms: β1, β2, β3) | Platelets, T lymphocytes, macrophages, monocytes, neutrophils, fibroblasts, keratinocytes | Stimulates angiogenesis Stimulates leukocyte chemotaxis TGF-β1 stimulates wound matrix production (fibronectin, collagen glycosaminoglycans); regulation of inflammation TGF-β3 inhibits scar formation |
Insulin-like growth factors (IGF-1, IGF-2) | Platelets (IGF-1 in high concentrations in liver; IGF-2 in high concentrations in fetal growth); likely the effector of growth hormone action | Promote protein/extracellular matrix synthesis Increase membrane glucose transport |
Vascular endothelial growth factor | Macrophages, fibroblasts, endothelial cells, keratinocytes | Mitogen for endothelial cells (not fibroblasts) Stimulates angiogenesis Proinflammatory |
IL-1 IL-4 IL-6 Activin Angiopoitein-1/-2 CX3CL1 | Macrophages, leukocytes, keratinocytes, fibroblasts Leukocytes Fibroblasts, endothelial cells, macrophages, keratinocytes Keratinocytes, fibroblasts Endothelial cells Macrophages, endothelial cells | Proinflammatory Stimulates angiogenesis, re-epithelialization, tissue remodeling Enhances collagen synthesis Stimulates inflammation, angiogenesis, re-epithelialization, collagen deposition, tissue remodeling Stimulates granulation tissue formation, keratinocyte differentiation, re-epithelialization Stimulates angiogenesis Stimulates inflammation, angiogenesis, collagen deposition |
Granulocyte- macrophage colony- stimulating factor | Macrophage/monocytes, endothelial cells, fibroblasts | Stimulates macrophage differentiation/proliferation |
Growth factors act on cells via surface receptor binding. Various receptor types have been described, such as ion channels, G-protein linked, or enzyme linked. The response elicited in the cell is usually one of phosphorylation or dephosphorylation of second-messenger molecules through the action of phosphatases or kinases, resulting in activation or deactivation of proteins in the cytosol or nucleus of the target cell. Phosphorylation of nuclear proteins is followed by the initiation of transcription of target genes.26 The signal is stopped by internalization of the receptor-ligand complex.
All wounds undergo some degree of contraction. For wounds that do not have surgically approximated edges, the area of the wound will be decreased by this action (healing by secondary intention); the shortening of the scar itself results in contracture. The myofibroblast has been postulated as being the major cell responsible for contraction, and it differs from the normal fibroblast in that it possesses a cytoskeletal structure. Typically this cell contains α-smooth muscle actin in thick bundles called stress fibers, giving myofibroblasts contractile capability.27 The α-smooth muscle actin is undetectable until day 6, and then is increasingly expressed for the next 15 days of wound healing.28 After 4 weeks, this expression fades and the cells are believed to undergo apoptosis.29 A puzzling point is that the identification of myofibroblasts in the wound does not correspond directly to the initiation of wound contraction, which starts almost immediately after injury.
Fibroblasts placed in a collagen lattice in vitro actively move in the lattice and contract it without expressing stress fibers. It is postulated that the movement of cells with concomitant reorganization of the cytoskeleton is responsible for contraction.30
HERITABLE DISEASES OF CONNECTIVE TISSUE
Heritable diseases of connective tissue consist of a group of generalized, genetically determined, primary disorders of one of the elements of connective tissue: collagen, elastin, or mucopolysaccharide. Five major types, Ehlers-Danlos syndrome, Marfan’s syndrome, osteogenesis imperfecta, epidermolysis bullosa, and acrodermatitis enteropathica, will be discussed, as each provides unique challenges to the surgeon.
Ehlers-Danlos syndrome (EDS) is a group of 10 disorders that present as a defect in collagen formation. Over half of the affected patients manifest genetic defects encoding alpha chains of collagen type V, causing it to be either quantitatively or structurally defective. These changes lead to “classic” EDS with phenotypic findings that include thin, friable skin with prominent veins, easy bruising, poor wound healing, atrophic scar formation, recurrent hernias, and hyperextensible joints. Gastrointestinal problems include bleeding, hiatal hernia, intestinal diverticulae, and rectal prolapse. Small blood vessels are fragile, making suturing difficult during surgery. Large vessels may develop aneurysms, varicosities, or arteriovenous fistulas or may spontaneously rupture.31,32,33 Table 9-3 presents a description of EDS subtypes including a recently recognized autosomal recessive form characterized by tenascin-X deficiency. The defect is a quantitative loss of protein, resulting in phenotypic changes similar to those observed in other types of EDS.
TYPE | CLINICAL FEATURES | INHERITANCE | BIOCHEMICAL DEFECT |
|---|---|---|---|
I | Skin: soft, hyperextensible, easy bruising, fragile, atrophic scars; hypermobile joints; varicose veins; premature births | AD | Not known |
II | Similar to type I, except less severe | AD | Not known |
III | Skin: soft, not hyperextensible, normal scars; small and large joint hypermobility | AD | Not known |
IV | Skin: thin, translucent, visible veins, normal scarring, no hyperextensibility; no joint hypermobility; arterial, bowel, and uterine rupture | AD | Type III collagen defect |
V | Similar to type II | XLR | Not known |
VI | Skin: hyperextensible, fragile, easy bruising; hypermobile joints; hypotonia; kyphoscoliosis | AR | Lysyl hydroxylase deficiency |
VII | Skin: soft, mild hyperextensibility, no increased fragility; extremely lax joints with dislocations | AD | Type I collagen gene defect |
VIII | Skin: soft, hyperextensible, easy bruising, abnormal scars with purple discoloration; hypermobile joints; generalized periodontitis | AD | Not known |
IX | Skin: soft, lax; bladder diverticula and rupture; limited pronation and supination; broad clavicle; occipital horns | XLR | Lysyl oxidase defect with abnormal copper use |
X | Similar to type II with abnormal clotting studies | AR | Fibronectin defect |
TNx | Hypermobile joints, skin fragility | AR | Absence of tenascin X protein |
EDS must be considered in every child with recurrent hernias and coagulopathy, especially when accompanied by platelet abnormalities and low coagulation factor levels. Inguinal hernias in these children resemble those seen in adults. Great care should be taken to avoid tearing the skin and fascia. The transversalis fascia is thin, and the internal ring is greatly dilated. An adult-type repair with the use of mesh or felt may result in a lower incidence of recurrence.34
The biochemical changes and phenotypic manifestation of the disease represent a major challenge to the surgeon. Dermal wounds should be closed in two layers, approximated with the sutures under tension, and the stitches should be left in place twice as long as usual. In addition, external fixation with adhesive tape can help reinforce the scar and prevent stretching.35
Patients with Marfan’s syndrome have tall stature, arachnodactyly, lax ligaments, myopia, scoliosis, pectus excavatum, and aneurysm of the ascending aorta. Patients who suffer from this syndrome also are prone to hernias. Surgical repair of a dissecting aneurysm is difficult, as the soft connective tissue fails to hold sutures. Skin may be hyperextensible but shows no delay in wound healing.36,37
The genetic defect associated with Marfan’s syndrome is a mutation in the FBN1 gene, which encodes for fibrillin. Previously, it was thought that structural alteration of the microfibrillar system was responsible for the phenotypic changes seen with the disease. However, recent research indicates an intricate role that FBN1 gene products play in TGF-β signaling. These extracellular matrix molecules normally bind and regulate TGF-β signaling; abnormal FBN1 gene function may cause an increase in TGF-β signaling, particularly in the aortic wall.38
Patients with osteogenesis imperfecta (OI) have brittle bones, osteopenia, low muscle mass, hernias, and ligament and joint laxity. OI is a result of a mutation in type I collagen. Mutations in prolidase, an enzyme responsible for cleaving c-terminal proline and hydroxyproline, may have a role in the disease. There are four major OI subtypes with mild to lethal manifestations. Patients experience dermal thinning and increased bruisability. Scarring is normal, and the skin is not hyperextensible. Surgery can be successful but difficult in these patients, as the bones fracture easily under minimal stress.31,34 Table 9-4 lists the various features associated with the clinical subtypes of OI.
TYPE | CLINICAL FEATURES | INHERITANCE |
|---|---|---|
I | Mild bone fragility, blue sclera | Dominant |
II | “Prenatal lethal”; crumpled long bones, thin ribs, dark blue sclera | Dominant |
III | Progressively deforming; multiple fractures; early loss of ambulation | Dominant/recessive |
IV | Mild to moderate bone fragility; normal or gray sclera; mild short stature | Dominant |
Epidermolysis bullosa (EB) is classified into four major subtypes: EB simplex, junctional EB, dystrophic EB, and Kindler’s syndrome. The first three are determined by location in various skin layers; the last can present as multiple blisters throughout different layers of skin. There are identified genetic defects for each subtype, but the overall phenotype is remarkably similar. The disease manifestations include impairment in tissue adhesion within the epidermis, basement membrane, or dermis, resulting in tissue separation and blistering with minimal trauma. Characteristic features of EB are blistering and ulceration. The recessively inherited dystrophic type is characterized by defects in the COL7A1 gene, encoding type 7 collagen, important for connecting the epidermis to the dermis, and therefore phenotypically resulting in blistering.39 Management of nonhealing wounds in patients with EB is a challenge, as their nutritional status is compromised because of oral erosions and esophageal obstruction. Surgical interventions include esophageal dilatation and gastrostomy tube placement. Dermal incisions must be meticulously placed to avoid further trauma to skin.34,40 The skin requires nonadhesive pads covered by a “bulky” dressing to avoid blistering.
Acrodermatitis enteropathica (AE) is an autosomal recessive disease of children that causes an inability to absorb sufficient zinc from breast milk or food. The AE mutation affects zinc uptake in the intestine by preventing zinc from binding to the cell surface and its translocation into the cell. Recently, the genetic defect has been localized on chromosome 8q24.3 identified as the SLC39A4 gene, expressed in the intestinal lumen and upregulated based on zinc stores.41 Zinc deficiency is associated with impaired granulation tissue formation, as zinc is a necessary cofactor for DNA polymerase and reverse transcriptase, and its deficiency may impair healing due to inhibition of cell proliferation.
AE is characterized by impaired wound healing as well as erythematous pustular dermatitis involving the extremities and the areas around the bodily orifices. Diagnosis is confirmed by the presence of an abnormally low blood zinc level (>100 mg/dL). Oral supplementation with 100 to 400 mg zinc sulfate orally per day is curative for impaired healing.42,43
HEALING IN SPECIFIC TISSUES
Healing of full-thickness injury to the gastrointestinal (GI) tract remains an unresolved clinical issue. Healing of full-thickness GI wounds begins with a surgical or mechanical reapposition of the bowel ends, which is most often the initial step in the repair process. Sutures or staples are principally used, although various other means such as buttons, plastic tubes, and various wrappings have been attempted with variable success. Failure of healing results in dehiscence, leaks, and fistulas, which carry significant morbidity and mortality. Conversely, excessive healing can be just as troublesome, resulting in stricture formation and stenosis of the lumen. Repair of the GI tract is vital to restoring the integrity of the luminal structure and to the resumption of motor, absorptive, and barrier functions.
The gross anatomic features of the GI tract are remarkably constant throughout most of its length. Within the lumen, the epithelium is supported by the lamina propria and underlying muscularis mucosa. The submucosa lies radially and circumferentially outside of these layers, is comprised of abundant collagenous and elastic fibers, and supports neural and vascular structures. Further toward the peritoneal surface of the bowel are the inner and outer muscle layers and ultimately a peritoneal extension, the serosa. The submucosa is the layer that imparts the greatest tensile strength and greatest suture-holding capacity, a characteristic that should be kept in mind during surgical repair of the GI tract. Additionally, serosal healing is essential for quickly achieving a watertight seal from the luminal side of the bowel. The importance of the serosa is underscored by the significantly higher rates of anastomotic failure observed clinically in segments of bowel that are extraperitoneal and lack serosa (i.e., the esophagus and rectum).
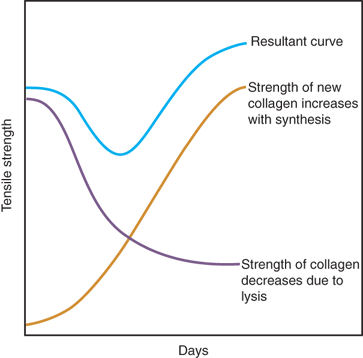
Injuries to all parts of the GI tract undergo the same sequence of healing as cutaneous wounds. However, there are some significant differences (Table 9-5). Mesothelial (serosal) and mucosal healing can occur without scarring. The early integrity of the anastomosis is dependent on formation of a fibrin seal on the serosal side, which achieves watertightness, and on the suture-holding capacity of the intestinal wall, particularly the submucosal layer. There is a significant decrease in marginal strength during the first week due to an early and marked collagenolysis. The lysis of collagen is carried out by collagenase derived from neutrophils, macrophages, and intraluminal bacteria. Recently, it has been shown that strains of Pseudomonas aeruginosa undergo phenotypic shifts characterized by higher collagenase secretion in an injured/anastomosed bowel environment.44 Collagenase activity occurs early in the healing process, and during the first 3 to 5 days, collagen breakdown far exceeds collagen synthesis. The integrity of the anastomosis represents equilibrium between collagen lysis, which occurs early, and collagen synthesis, which takes a few days to initiate (Fig. 9-5). Collagenase is expressed postinjury in all segments of the GI tract, but it is much more marked in the colon compared to the small bowel. Collagen synthesis in the GI tract is carried out by both fibroblasts and smooth muscle cells. Colon fibroblasts produce greater amounts of collagen than skin fibroblasts, reflecting different phenotypic features, as well as different responses to cytokines and growth factors among these different fibroblast populations. Ultimate anastomotic strength is not always related to the absolute amount of collagen, and the structure and arrangement of the collagen matrix may be more important.45
GI TRACT | SKIN | ||
|---|---|---|---|
Wound environment | pH | Varies throughout GI tract in accordance with local exocrine secretions | Usually constant except during sepsis or local infection |
Microorganisms | Aerobic and anaerobic, especially in the colon and rectum; problematic if they contaminate the peritoneal cavity | Skin commensals rarely cause problems; infection usually results from exogenous contamination or hematogenous spread | |
Shear stress | Intraluminal bulk transit and peristalsis exert distracting forces on the anastomosis | Skeletal movements may stress the suture line but pain usually acts as a protective mechanism preventing excess movement | |
Tissue oxygenation | Dependent on intact vascular supply and neocapillary formation | Circulatory transport of oxygen as well as diffusion | |
Collagen synthesis | Cell type | Fibroblasts and smooth muscle cells | Fibroblasts |
Lathyrogens | d-Penicillamine has no effect on collagen cross-linking | Significant inhibition of cross-linking with decreased wound strength | |
Steroids | Contradictory evidence exists concerning their negative effect on GI healing; increased abscess in the anastomotic line may play a significant role | Significant decrease in collagen accumulation | |
Collagenase activity | — | Increased presence throughout GI tract after transection and reanastomosis; during sepsis excess enzyme may promote dehiscence by decreasing suture-holding capacity of tissue | Not as significant a role in cutaneous wounds |
Wound strength | — | Rapid recovery to preoperative level. | Less rapid than GI tissue |
Scar formation | Age | Definite scarring seen in fetal wound sites | Usually heals without scar formation in the fetus |
Figure 9-5.
Diagrammatic representation of the concept of GI wound healing as a fine balance between collagen synthesis and collagenolysis. The “weak” period when collagenolysis exceeds collagen synthesis can be prolonged or exacerbated by any factors that upset the equilibrium. (Reproduced with permission from Hunt TK, Van Winkle W Jr. Wound healing: normal repair. In: Dunphy JE, ed. Fundamentals of Wound Management in Surgery. New York: Chirurgecom, Inc.; 1976:29.)
Traditional teaching holds that in order for an anastomosis to heal without complications it must be tension-free, have an adequate blood supply, receive adequate nutrition, and be free of sepsis. Although sound principles for all wound healing, there are several considerations unique to anastomotic healing. From a technical viewpoint, the ideal method of suturing two ends of bowel together has not yet been identified. Although debate exists concerning methods of creating an anastomosis, clinically there has been no convincing evidence that a given technique has any advantage over another (i.e., hand-sutured vs. stapled, continuous vs. interrupted sutures, absorbable vs. nonabsorbable sutures, or single- vs. two-layer closure). A recent meta-analysis revealed that stapled ileocolic anastomoses have fewer leak rates than hand-constructed ones, but no data on colo-colic or small bowel anastomoses have been offered yet.46 It is known, however, that hand-sutured everting anastomoses are at greater risk of leakage and cause greater adhesion formation, but have a lower incidence of stenosis. Because no overall definite superiority of any one method exists, it is recommended that surgeons be familiar with several techniques and apply them as circumstances dictate.
The amount of intravenous fluid administered perioperatively affects many aspects of recovery from colonic surgery; experimental and clinical data show that anastomotic healing may be adversely affected by overzealous fluid administration, which results in fluid accumulation in the third space, increased abdominal pressure, and tissue edema, all of which can compromise blood flow in the small vessels at the healing edge.47,48
Following any type of injury to bone, several changes take place at the site of injury to restore structural and functional integrity. Most of the phases of healing resemble those observed in dermal healing, but some notable individual characteristics apply to bone injuries. The initial stage of hematoma formation consists of an accumulation of blood at the fracture site, which also contains devitalized soft tissue, dead bone, and necrotic marrow. The next stage accomplishes the liquefaction and degradation of nonviable products at the fracture site. The normal bone adjacent to the injury site can then undergo revascularization, with new blood vessels growing into the fracture site. This is similar to the formation of granulation tissue in soft tissue. The symptoms associated with this stage are characteristic of inflammation, with clinical evidence of swelling and erythema.
Three to 4 days following injury, soft tissue forms a bridge between the fractured bone segments in the next stage (soft callus stage). This soft tissue is deposited where neovascularization has taken place and serves as an internal splint, preventing damage to the newly laid blood vessels and achieving a fibrocartilaginous union. The soft callus is formed externally along the bone shaft and internally within the marrow cavity. Clinically, this phase is characterized by the end of pain and inflammatory signs.
The next phase (hard callus stage) consists of mineralization of the soft callus and conversion to bone. This may take up to 2 to 3 months and leads to complete bony union. The bone is now considered strong enough to allow weight bearing and will appear healed on radiographs. This stage is followed by the remodeling phase, in which the excessive callus is reabsorbed and the marrow cavity is recanalized. This remodeling allows for the correct transmission of forces and restores the contours of the bone.
As in dermal healing, the process of osseous union is mediated by soluble growth factors and cytokines. The most extensively studied group is the bone morphogenic proteins (BMPs), which belong to the TGF-β superfamily. By stimulating the differentiation of mesenchymal cells into chondroblasts and osteoblasts, BMPs directly affect bone and cartilage repair. Other growth factors such as PDGF, TGF-β, TNF-α, and bFGF also participate in bony repair by mediating the inflammatory and proliferative phases of healing.
Cartilage consists of cells (chondrocytes) surrounded by an extracellular matrix made up of several proteoglycans, collagen fibers, and water. Unlike bone, cartilage is very avascular and depends on diffusion for transmittal of nutrients across the matrix. Additionally, the hypervascular perichondrium contributes substantially to the nutrition of the cartilage. Therefore, injuries to cartilage may be associated with permanent defects due to the meager and tenuous blood supply.
The healing response of cartilage depends on the depth of injury. In a superficial injury, there is disruption of the proteoglycan matrix and injury to the chondrocytes. There is no inflammatory response, but an increase in synthesis of proteoglycan and collagen dependent entirely on the chondrocyte. Unfortunately, the healing power of cartilage is often inadequate, and overall regeneration is incomplete. Therefore, superficial cartilage injuries are slow to heal and often result in persistent structural defects.
In contrast to superficial injuries, deep injuries involve the underlying bone and soft tissue. This leads to the exposure of vascular channels of the surrounding damaged tissue that may help in the formation of granulation tissue. Hemorrhage allows for the initiation of the inflammatory response and the subsequent mediator activation of cellular function for repair. As the granulation tissue is laid down, fibroblasts migrate toward the wound and synthesize fibrous tissue that undergoes chondrification. Gradually, hyaline cartilage is formed, which restores the structural and functional integrity of the injured site.
Tendons and ligaments are specialized structures that link muscle and bone, and bone and bone, respectively. They consist of parallel bundles of collagen interspersed with spindle cells. Tendons and ligaments can be subjected to a variety of injuries, such as laceration, rupture, and contusion. Due to the mobility of the underlying bone or muscles, the damaged ends usually separate. Tendon and ligament healing progresses in a similar fashion as in other areas of the body (i.e., through hematoma formation, organization, laying down of reparative tissue, and scar formation). Matrix is characterized by accumulation of type I and III collagen along with increased water, DNA, and glycosaminoglycan content. As the collagen fibers are organized, transmission of forces across the damaged portion can occur. Restoration of the mechanical integrity may never be equal to that of the undamaged tendon.
Tendon vasculature has a clear effect on healing. Hypovascular tendons tend to heal with less motion and more scar formation than tendons with better blood supply. The specialized cells, tenocytes, are metabolically very active and retain a large regenerative potential, even in the absence of vascularity. Cells on the tendon surface are identical to those within the sheath and play a role in tendon healing as well.
Nerve injuries are very common, with an estimated 200,000 repairs performed every year in the United States. Peripheral nerves are a complex arrangement of axons, nonneuronal cells, and extracellular elements. There are three types of nerve injuries: neurapraxia (focal demyelination), axonotmesis (interruption of axonal continuity but preservation of Schwann cell basal lamina), and neurotmesis (complete transection). Following all types of injury, the nerve ends progress through a predictable pattern of changes involving three crucial steps: (a) survival of axonal cell bodies; (b) regeneration of axons that grow across the transected nerve to reach the distal stump; and (c) migration and connection of the regenerating nerve ends to the appropriate nerve ends or organ targets.
Phagocytes remove the degenerating axons and myelin sheath from the distal stump (Wallerian degeneration). Regenerating axonal sprouts extend from the proximal stump and probe the distal stump and the surrounding tissues. Schwann cells ensheathe and help in remyelinating the regenerating axons. Functional units are formed when the regenerating axons connect with the appropriate end targets. Several factors play a role in nerve healing, such as growth factors, cell adhesion molecules, and nonneuronal cells and receptors. Growth factors include nerve growth factor, brain-derived neurotrophic factor, basic and acidic fibroblastic growth factors, and neuroleukin. Cell adhesion molecules involved in nerve healing include nerve adhesion molecule, neuron-glia adhesion molecule, myelin adhesion glycoprotein, and N-cadherin. This complex interplay of growth factors and adhesion molecules helps in nerve regeneration.
The main characteristic that distinguishes the healing of fetal wounds from that of adult wounds is the lack of scar formation. Understanding how fetal wounds achieve integrity without evidence of scarring holds promise for the possible manipulation of unwanted fibrosis or excessive scar formation in adults.
Although early fetal healing is characterized by the absence of scarring and resembles tissue regeneration, there is a phase of transition during gestational life when a more adult-like healing pattern emerges. This so-called “transition wound” occurs at the beginning of the third trimester, and during this period, there is scarless healing; however, there is a loss of the ability to regenerate skin appendages.49 Eventually a classic, adult-patterned healing with scar formation occurs exclusively, although overall healing continues to be faster than in adults.
There are a number of characteristics that may influence the differences between fetal and adult wounds. These include wound environment, inflammatory responses, differential growth factor profiles, and wound matrix.
The fetus is bathed in a sterile, temperature-stable fluid environment, although this alone does not explain the observed differences. Experiments have demonstrated that scarless healing may occur outside of the amniotic fluid environment, and conversely, scars can form in utero.50,51



