Usual Interstitial Pneumonia
Key Facts
Terminology
Pattern of lung damage characterized by bilateral, diffuse, interstitial inflammation and fibrosis
Clinical Issues
Insidious onset of dyspnea and nonproductive cough
Poor prognosis
Mean survival ranges from 3.5-5 years
Image Findings
HRCT shows reticular pattern with honeycombing involving mainly subpleural lung regions
In more advanced stages there is traction bronchiectasis and honeycombing
Macroscopic Features
Peripheral subpleural areas of fibrosis of lung parenchyma predominantly in lower lobes
Peripheral honeycomb cystic changes in more advanced lesions
Microscopic Pathology
Areas of lung parenchyma showing interstitial inflammation and fibrosis adjacent to areas of normal parenchyma
Areas of diseased lung are seen in various stages of evolution (“temporal heterogeneity”)
Interstitial fibrosis causes widening of alveolar septa due to collagen deposition admixed with inflammatory cells
“Fibroblastic foci” are present, composed of loose connective tissue admixed with fibroblastic cells
Areas of fibrosis with cystic spaces result in “honeycombing” effect
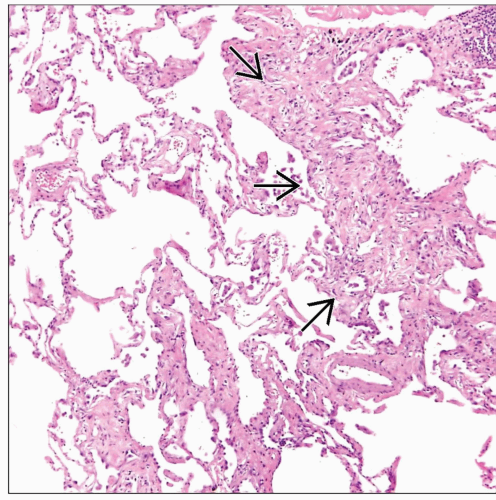 Scanning magnification in usual interstitial pneumonia shows areas of lung parenchyma with thickened alveolar walls  surrounded by areas of normal lung parenchyma. surrounded by areas of normal lung parenchyma. |
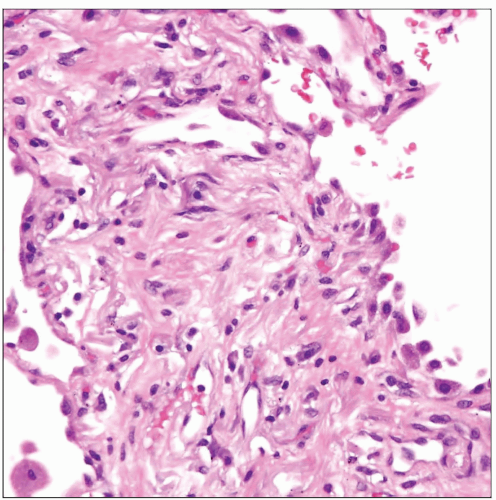 Higher magnification of thickened alveolar wall in usual interstitial pneumonia shows heavy deposition of collagen fibers admixed with sparse inflammatory infiltrate. |
TERMINOLOGY
Abbreviations
Usual interstitial pneumonia (UIP)
Synonyms
Idiopathic interstitial fibrosis, idiopathic pulmonary fibrosis, cryptogenic fibrosing alveolitis
Definitions
Pattern of lung damage characterized by bilateral, diffuse, interstitial inflammation and fibrosis
ETIOLOGY/PATHOGENESIS
Pathogenesis
Unknown (idiopathic)
May be associated with a variety of clinical conditions, including
Collagen-vascular disease
Drug toxicity
Pneumoconiosis and other environmental exposures
CLINICAL ISSUES
Epidemiology
Incidence
Approximately 10 cases per 100,000 people per year
Age
50-70 years of age
Gender
More common in men
Presentation
Insidious onset of dyspnea and nonproductive cough
Tachypnea
Bibasilar, late inspiratory crackles
Clubbing of fingers in > 40% of patients
Pulmonary hypertension (in late stages)
Restriction and impairment of gas exchange on pulmonary function tests
Treatment
Surgical approaches
Lung transplantation
Drugs
Immunosuppressive and cytotoxic agents have not been very effective
Gamma interferon
Prognosis
Poor prognosis
Mean survival ranges from 3.5-5 years
Respiratory failure is most frequent cause of death
IMAGE FINDINGS
Radiographic Findings
Bilateral, symmetrical, linear opacities showing reticular pattern
Ground-glass opacities
Honeycombing and decreased lung volume
Abnormalities involve mainly lower lobes of lung
Normal chest x-rays may be seen in ˜ 10% of patients
CT Findings
HRCT shows reticular pattern with honeycombing involving mainly subpleural lung regions
Intralobular linear opacities with irregular thickening of interlobular septa
Irregular pleural, vascular, and bronchial interfaces with the lung parenchyma
In more advanced stages there is traction bronchiectasis and honeycombing
Typical findings may be seen on HRCT even when chest x-rays appear normal
Bilateral process with basilar and peripheral predominance
MACROSCOPIC FEATURES
General Features
Peripheral subpleural areas of fibrosis of lung parenchyma, predominantly in lower lobes
Peripheral honeycomb cystic changes in more advanced lesions
MICROSCOPIC PATHOLOGY
Histologic Features
Areas of lung parenchyma showing interstitial inflammation and fibrosis adjacent to areas of normal parenchyma
Areas of diseased lung are seen in various stages of evolution (“temporal heterogeneity”)
Interstitial fibrosis causes widening of alveolar septa due to collagen deposition admixed with inflammatory cells
Interstitial inflammatory infiltrate includes small lymphocytes, plasma cells, and histiocytes with occasional lymphoid follicles
“Fibroblastic foci” are present, composed of loose connective tissue admixed with fibroblastic cells
Focal accumulation of alveolar macrophages (“DIP-like” appearance)
Bronchiolization of alveolar lining (“Lambertosis”) may be observed
Squamous metaplasia in areas of scarring can be seen in late stages
Smooth muscle stromal proliferation may accompany stromal scarring
Areas of fibrosis with cystic spaces result in “honeycombing” effect
Episodes of acute exacerbation may result in foci of diffuse alveolar damage superimposed on UIP
DIAGNOSTIC CHECKLIST
Clinically Relevant Pathologic Features
Transbronchial biopsies are not useful for evaluation of interstitial fibrosis
Multiple open lung biopsies are indicated for definitive diagnosis
Biopsies must be obtained from multiple lobes, avoiding the lung apices
About 50% of patients can be diagnosed on clinical and radiologic grounds alone without a biopsy
Pathologic Interpretation Pearls
“Temporal heterogeneity” (lesions at various stages of evolution) serve to distinguish UIP from NSIP
Clinical history is indispensable for cases associated with underlying collagen-vascular disorders
Scanning magnification showing variegation and admixture of abnormal foci with normal lung parenchyma is diagnostic
Advanced stages with extensive fibrosis and honeycombing may be impossible to diagnose in absence of a history
SELECTED REFERENCES
1. Borchers AT et al: Idiopathic Pulmonary Fibrosis-an Epidemiological and Pathological Review. Clin Rev Allergy Immunol. 40(2):117-34, 2011
2. Chan AL et al: Therapeutic Update in Idiopathic Pulmonary Fibrosis. Clin Rev Allergy Immunol. Epub ahead of print, 2011
3. Force SD et al: Bilateral lung transplantation offers better long-term survival, compared with single-lung transplantation, for younger patients with idiopathic pulmonary fibrosis. Ann Thorac Surg. 91(1):244-9, 2011
4. Cavazza A et al: The role of histology in idiopathic pulmonary fibrosis: an update. Respir Med. 104 Suppl 1:S11-22, 2010
5. Coward WR et al: The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 4(6):367-88, 2010



