Discovery
Potential mechanisms of action
Range of complications
TABLE 5-1 ANTIPSYCHOTIC AGENTS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
effects can be dissociated. Initially, the term neuroleptic was applied to all drugs that produce both extrapyramidal and antipsychotic effects, but agents such as clozapine, RISP, OLZ, QTP, ZPD, ARIP, and others can be antipsychotics without being neuroleptics, making the interchangeable use of the terms neuroleptic and antipsychotic no longer appropriate.
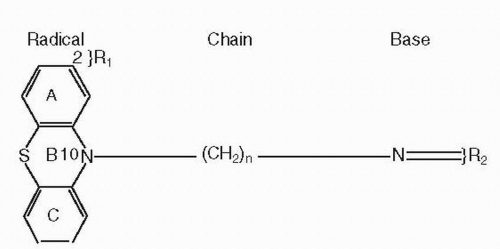 Figure 5-1 Basic requirements for antipsychotic activity with first-generation antipsychotics (FGAs); distance between N10 of radical and the base N atom must equa at least three carbons (n = 3), with a suitable substituent R2 mainly determining the qualitative properties of the group; the A2 substituent R1 mainly determines the quantitative properties of the individual compounds. |
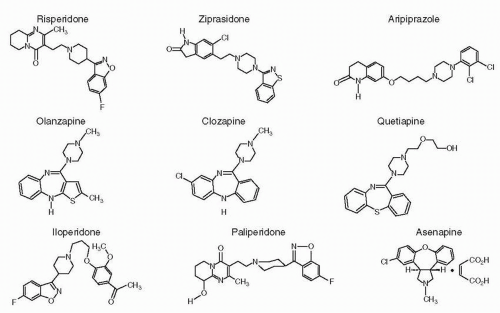 Figure 5-2 Chemical structures of second-generation antipsychotics (SGAs). |
psychosis, it is pertinent to discuss the central pathways of this neurotransmitter. There are five tracts:
The striatal system (A-9)
The mesolimbic system (A-10)
The mesocortical system (A-10)
The retinal system
The neurohypophyseal system
Activate tyrosine hydroxylase in the presynaptic dopaminergic neuron
Increase DA synthesis
Increase the firing rate
Increase the levels of DA metabolites such as homovanillic acid (HVA)
inducing paranoia, hallucinations, and other manifestations of psychosis. For example,
L-Dopa (3,4-dihydroxyphenylalanine), which is converted to DA in the body, can produce psychosis as a side effect
Amantadine, another dopaminergic drug, can also induce psychotic symptoms
Stimulants, such as amphetamine and methylphenidate, are potent releasers of DA, whereas cocaine also interferes with its reuptake, increasing DA concentrations at synaptic sites; all can produce paranoid reactions in some abusers
Bromocriptine, apomorphine, lisuride, and other direct-acting DA agonists benefit Parkinson’s disease and can also cause psychotic reactions at high doses
Serotonin (5-HT)
Norepinephrine (NE)
γ-Aminobutyric acid (GABA)
Glutamate (GLU)
Neurohormones
Neuropeptides
ARIP) are partial agonists at the 5-HT1A receptor, which may also modulate DA activity.
TABLE 5-2 NEURORECEPTOR BINDING PROFILE OF SECOND-GENERATION ANTIPSYCHOTICSa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Good absorption from the gastrointestinal tract
An extensive “first-pass” hepatic effect
Subsequent high systemic clearance due to a large hepatic extraction ratio each time the plasma recirculates through the liver
Extensive distribution (VD) due to highly lipophilic character
Plasma half-life (t1/2) of typically about 20 hours
Primary route of elimination through hepatic metabolism (except PALI)
The presence of metabolites with varying pharmacological profiles (e.g., some may be more effective than their parent compound [e.g., mesoridazine], some may not reach the brain [e.g., sulfoxides], and some may have greater toxicity than the parent compound).
The existence of a long interval between drug administration and clinical response
Large interindividual differences in response to the same dose for the same diagnosis, in part reflecting differences in plasma levels achieved on a given dose
Possible usefulness in helping to establish the minimally effective dose
Possible usefulness in helping to estimate the average dose required to achieve a certain concentration when a positive correlation exists between a given steady state concentration (Css) and the dose required.
Insufficient sample sizes, especially at the low and the high ranges
Inclusion of refractory, nonhomogeneous, nonadherent patients
Nonrandom adjustment of the dose based on response, adverse effects, or initial presentation
Concurrent treatments
Too brief an observation period for clinical effects to occur
Variable time of blood sampling
Inadequate evaluation of clinical response as well as the indefinite nature of the response
Presence of numerous active metabolites
Inadequate assay methods, such as radioreceptor assays
in the brain at levels about 10 to 30 times higher than the plasma level. Animal studies find a brain-to-plasma ratio of about 20:1. Limited information from animal studies after drug withdrawal also shows a more rapid disappearance from plasma than from the brain. Although the half-life of HPDL is about 24 hours in plasma, even several days after cessation of oral dosing, significant CNS D2 receptor activity is still evident. Indeed, PET studies indicate that after withdrawal of HPDL or fluphenazine decanoate, D2 blockade can persist for several months. Thus, it is likely that human brain levels are approximately 10 to 20 times higher than plasma levels and that these agents stay in the brain longer than in plasma.
TABLE 5-3 PHARMACOKINETICS OF SECOND-GENERATION ANTIPSYCHOTICS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
that 9 to 15 mg of trifluoperazine would be comparable to 300 mg of CPZ and should fall near the lower part of the linear portion of the dose-response curve.
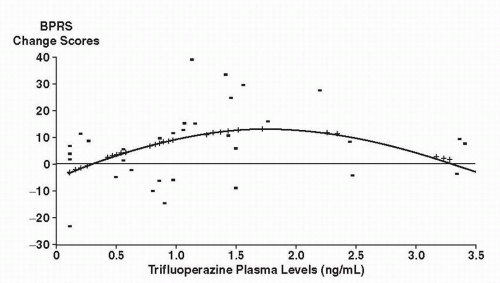 Figure 5-3 Brief Psychiatric Rating Scale (BPRS) change scores in relationship to trifluoperazine plasma levels. (From Janicak PG, Javaid JI, Sharma RP, et al. Trifluoperazine plasma levels and clinical response. J Clin Psychopharmacol. 1989;9:340-346, with permission.) |
treatment to yield a distribution of plasma levels above and below a hypothesized therapeutic level of 18 ng/mL. They found a significant negative linear relationship (i.e., as dose increased, response decreased) between HPDL plasma levels and outcome at week 1, which did not persist into weeks 2, 3, and 4.
TABLE 5-4 EARLY FIXED-DOSE STUDIES FINDING A CURVILINEAR RELATIONSHIP BETWEEN HALOPERIDOL PLASMA LEVELS AND CLINICAL RESPONSE | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
as the reason for improvement, especially when the dose and blood levels were reduced in the first patient.
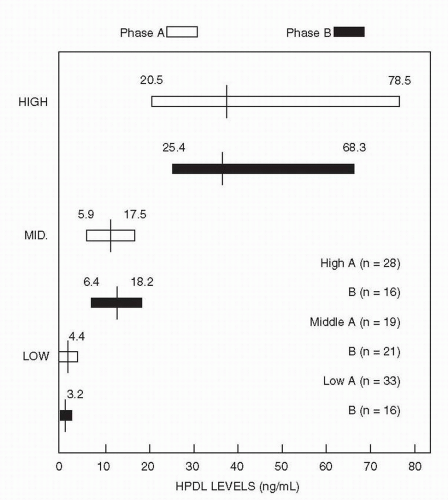 Figure 5-4 Haloperidol (HPDL) plasma level ranges by targeted groups. (Adapted from Janicak PG, Javaid JI, Sharma RP, et al. A two-phase, double-blind randomized study of three haloperidol blood levels for acute psychosis with reassignment of initial non-responders. Acta Psychiatr Scand. 1997;95:343-350.) |
time points, the Janicak et al. (63) study found a greater improvement in phase B (8.1 BPRS points) compared with McEvoy et al.’s second phase (2.9 BPRS change score points). In the McEvoy study, although higher doses of HPDL did not produce greater improvement in measures of psychosis, they did lead to slightly more improvement in hostility.
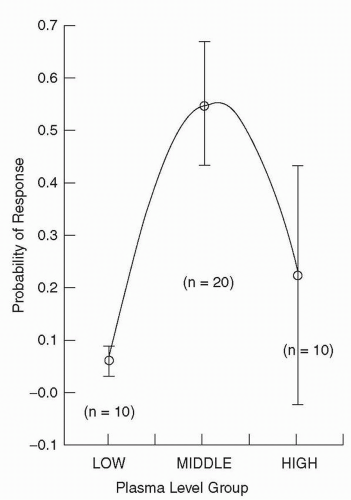 Figure 5-5 Clinical response based on plasma leve l assignment in Phase B. (From Janicak PG, Javaid JI, Sharma RP, et al. A two-phase, double-blind randomized study of three haloperidol plasma levels for acute psychosis with reassignment of initial non-responders. Acta Psychiatr Scand. 1997;95:343-350, with permission.) |
additional benefit to using higher doses or plasma levels.
identified 350 ng/mL as a threshold for response. At or above this plasma level, 64% of patients responded as compared with only 23% of patients below that level. This group followed up these original results with a larger sample, and in a follow-up of the existing samples, five of seven patients had an unsatisfactory response to a dose of 400 mg/day. When the dose was increased to achieve a plasma level above the threshold 350 ng/mL, however, most then responded (76).
Determine patient adherence
Establish that adequate doses of pharmacotherapy were used in nonresponders
Avoid toxicity due to unnecessarily high plasma levels
Monitor patients with other medical disorders or on concurrent psychotropics or other medical drugs
Maximize the clinical response when the drug plasma level-response relationship has been elucidated
Help define the dose-response relationship
Safeguard clinicians in potential medicolegal situations
TABLE 5-5 EFFICACY OF ANTIPSYCHOTICS | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
16% of the drug-treated versus only 1% of the placebo-treated group were in complete remission or very much improved
29% of the drug-treated and 11% of the placebo-treated group were evaluated as improved, with only borderline symptoms
16% of the drug-treated and 10% of the placebo-treated group were evaluated as improved, with mild symptoms remaining
8% of patients did poorly on active drug (i.e., rated as not improved or still ill), compared with 48% on placebo
Only 2% of those on an antipsychotic versus 33% of the placebo patients deteriorated during 6 weeks of treatment
The largest differences between the drug- and the placebo-treated patients are seen at both ends of the outcome spectrum
studies that compared these SGAs with both a placebo and an FGA, usually HPDL. All of these early studies found that the SGA was superior to placebo, and some found that the SGA was also more effective than HPDL for psychotic symptoms. However, a broader look at these and studies of other SGAs raised questions about their advantages. A meta-analysis by Geddes et al. (88) concluded that the advantages of SGAs were apparent only when patients received excessive doses of the comparator FGA. More specifically, studies that used doses of HPDL greater than 12 mg daily were more likely to find advantages for the SGAs. This suggests that the greater severity of extrapyramidal symptoms in the HPDL-treated groups may have given the SGAs an apparent advantage, though it was not related to their intrinsic antipsychotic properties. In addition, another meta-analysis (87) indicated that even when there were statistically significant advantages for the newer drug over HPDL, the effect sizes were very small. This finding is disputed in a meta-analysis by Davis et al. (89) who found substantial advantages for clozapine, amisulpride, RISP, and OLZ when compared with HPDL. These advantages were not present with other SGAs. Cochrane reviews of RISP (90) and OLZ (91), however, point out design flaws such as high dropout rates that make it difficult to determine whether these SGAs are superior to FGAs.
Time to discontinuation for any cause was longer for OLZ (but this was nonsignificant for perphenazine and ZPD after adjustment for multiple comparisons)
Time to discontinuation for lack of efficacy was longer for OLZ (but this was nonsignificant for ZPD after adjustment for multiple comparisons)
Duration of successful treatment was significantly longer for OLZ
Time to discontinuation for intolerable side effects did not differ, but rates were highest in the OLZ group and lowest in the RISP group
Metabolic issues were highest with OLZ, while ZPD was the only agent associated with improvement in metabolic variables
EPSs were highest with perphenazine
Hyperprolactinemia only occurred with RISP
Clinically relevant changes in the QTc interval did not occur with any agent
TABLE 5-6 SCHIZOPHRENIA: SYMPTOM DIMENSIONS | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
although their effects are relatively weak compared with the severity of the impairments. Although a number of studies suggest that SGAs are superior to FGAs in improving cognition (99), patients treated with the newer agents usually still retain these impairments. For example, it is estimated that individuals with schizophrenia perform one and one half to two standard deviations below the mean on neuropsychological tests, and SGAs may improve performance by about one half of a standard deviation (99). Although a number of studies have compared SGAs to one another, it is unclear if any SGA demonstrates advantages over others for neurocognitive impairment (100). The NIMH CATIE study compared the effects of five different antipsychotics on a neurocognitive battery (101). Each of the medications led to relatively small improvements in a composite score (z = 0.12 to 0.26) after 2 months. Moreover, there were no differences among the antipsychotics which included perphenazine, an FGA. Interestingly, at 18 months, there was a suggestion that perphenazine had an advantage when compared with the SGAs. This provides further evidence that SGAs do not have specific effects on neurocognition.
and 67% (108,109). Thus, the low incidence of EPS with clozapine may be due to a subthreshold level of D2 occupancy at clinical doses.
Has a lower affinity for D2 receptors
Binds proportionately more to D1 and D4 receptors
Appears to act selectively on cortical and mesolimbic DA neuronal systems (in preference to the nigrostriatal and tuberoinfundibular areas)
Blocks 5-HT2 receptors
Has few or no EPS effects across the recommended dosing range
Has effects on several other neurotransmitter systems (e.g., NE, ACh)
Has more than 25 years of experience establishing it as an effective treatment for
Acute and chronic schizophrenia
Psychoses that are responsive or nonresponsive to other antipsychotics
the Mount Sinai Hospital in 2001 (242) also state that patients should not be considered partial or nonresponders until they have received a trial of clozapine. Conley et al. (123) underlined the importance of trying clozapine in patients who are doing poorly on another SGA when they found that individuals who failed to respond to OLZ did substantially better with clozapine.
Farren et al. (138), who administered ascending doses of clozapine followed by intranasal cocaine in addicted patients. Clozapine was reported to diminish the subjective “high” following the cocaine.
RISP, 9-OH-RISP, ZPD, and ARIP had the highest affinity for human D2 receptors
OLZ was slightly less potent at D2 receptor sites
RISP, 9-OH-RISP, OLZ, and clozapine maintained a higher potency for occupying 5-HT2A than D2 receptors
RISP had a 5-HT2A versus D2 potency ratio of about 20
High potency for 5-HT2A occupancy was observed for RISP and OLZ
achieves a D2 receptor occupancy similar to that of FGAs and higher than that of clozapine. The occurrence of EPS at higher levels of D2 receptor occupancy in this study and in previous clinical trials indicates that the high 5-HT2 affinity of RISP provides some protection against EPS. Clinical and laboratory data, however, also suggest that when the D2 receptor occupancy exceeds a certain threshold, any 5-HT2-mediated protection against EPS may be lost. The lower propensity of RISP to evoke EPS is progressively diminished as its dose increases and its occupancy of D2 receptors rises. Thus, there is a dosage range between inducing and not inducing EPS.
Schizophrenia in adults and adolescents
Bipolar mania in adults, children, and adolescents
Irritability associated with autistic disorder
Schizophrenia
As monotherapy or an adjunctive therapy to VPA or lithium for bipolar I disorder
and rapid mood change. Patients should be started on 0.25 mg/day for children weighing less than 20 kg and 0.5 mg/day for heavier children. Most patients respond between 0.5 and 2.5 mg daily.
Tic disorders
Mental retardation or pervasive developmental disorders
Autistic disorder
Psychiatric disorders caused by a medical condition (e.g., acquired immunodeficiency syndrome [AIDS]; organic delusional disorders)
Agitated or aggressive states, including those associated with dementia (NB SGAs, including RISP, now carry a recently mandated box warning for increased risk of cerebrovascular AEs and a box warning for increased mortality risk in patients with dementia and psychosis; see also Chapter 15) (159,160); further, the risk of death in elderly patients (>65 years) may be even higher on FGAs versus SGAs (161)
Anorexia nervosa
Personality disorders (e.g., borderline and schizotypal)
Obsessive-compulsive disorder
Posttraumatic stress disorder
Various other psychotic disorders, such as brief psychotic disorder, delusional disorder, shared psychotic disorder, substance-induced psychotic disorder, and psychotic disorder not otherwise specified (162)
found an advantage for RISP over placebo for irritability, insecurity/anxiety, and hyperactivity. RISP-treated patients, however, experienced greater sedation and weight gain. Snyder et al. (171) compared RISP and placebo in 110 children with IQs between 36 and 84 who exhibited conduct problems. The RISP-treated children demonstrated greater improvements than children on placebo on scales measuring the severity of the conduct problems.
The adverse effects of clozapine
The frequent venipunctures required for hematological monitoring
The cost of clozapine and laboratory testing
The hope that RISP will be safer and at least as effective as clozapine
addition of RISP to clozapine was well tolerated and produced significant reduction of symptoms on the total BPRS and the psychotic, negative symptom, and depression subscales. No patients complained of dizziness or demonstrated changes in their blood pressure or pulse.
Acute treatment for manic or mixed episodes in bipolar I disorder in adults and adolescents (also as an adjunct to VPA or lithium)
Maintenance treatment of bipolar I disorder
binding sites and causes selective activation of the A-10 DA neurons when given acutely and depolarization inactivation of A-10 DA neurons when given chronically (195). Early PET studies of QTP receptor occupancy indicated substantially lower D2 occupancy than with other SGAs, with the exception of clozapine, which is also associated with lower occupancy (196). Kapur et al. (13) found that QTP led to transiently high D2 occupancy of 58% to 64% during the first 2 to 3 hours, which fell to minimal levels by 12 hours. The extended and immediate release formulations of QTP result in similar plasma levels and D2 occupancy (197). These studies suggest that transient occupancy may be sufficient for an antipsychotic effect. These observations provide strong support for Seeman’s proposal (106) that an important property of SGAs is their ability to occupy D2 receptors briefly and then rapidly dissociate from them to allow normal DA neurotransmission.
Acute treatment for manic or mixed episodes in bipolar I disorder in adults and adolescents (also as an adjunct to VPA or lithium)
Depressed episodes in bipolar I and II disorder
Maintenance treatment of bipolar I disorder
Adjunctive treatment in major depressive disorder
of QTP (Seroquel XR) demonstrates similar efficacy when compared with twice-daily administration of the immediate release formulation (203,206).
Is a moderately potent D2 receptor antagonist (K; = 8.32)
Has a high affinity for the D3 receptor (pK. = 8.14)
Shows a 100-fold lower affinity for the D1 receptor (approximately equivalent to that of RISP and HPDL) than for the other DA receptor subtypes
Schizophrenia
Acute treatment as monotherapy for bipolar I manic or mixed episodes
Maintenance treatment of bipolar I disorder as an adjunct to lithium or VPA
As an intramuscular injection for the acute treatment of agitation in schizophrenia patients
TABLE 5-7 ZIPRASIDONE VERSUS PLACEBO OR HALOPERIDOL FOR ACUTE TREATMENT OF SCHIZOPHRENIA OR SCHIZOAFFECTIVE DISORDER | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Schizophrenia in adults and adolescents (ages 13 to 17 years)
Acute treatment of manic or mixed episodes associated with bipolar I disorder as monotherapy and as an adjunct to lithium or VPA
Maintenance treatment of bipolar I disorder
Treatment of irritability associated with autistic disorder
Acute and maintenance treatment of schizophrenia
Acute treatment of schizoaffective disorder as monotherapy
Acute treatment of schizoaffective disorder as an adjunct to mood stabilizers and/or antidepressants
PALI which is substantially more expensive when compared with generic RISP.
Acute treatment of schizophrenia in adults
Acute treatment of manic or mixed episodes in bipolar I disorder in adults
Because it may prolong the QT interval, other antipsychotics should be considered first.
There is a need to titrate the dose due to orthostatic hypotension. This may delay the onset of effectiveness.
underlying disease process. The result is that patients may enjoy a robust antipsychotic effect for positive symptoms but still retain psychopathology in other domains such as negative symptoms or cognitive impairments. Although goals may include eliminating every trace of hallucinations or delusions, even a substantial improvement in these positive symptoms may be associated with reduced suffering, improved functioning, and a better quality of life.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


