Learning Objectives
Define therapeutic drug monitoring, and learn when it is necessary and how it is performed for commonly monitored drugs.
Describe basic pharmacokinetic principles as they relate to therapeutic drug monitoring.
Identify the common drugs of abuse and how they are detected in blood, serum, urine, and other body fluids.
Understand the association between occupations, industries, and exposure to specific environmental toxins.
Introduction
Toxicology comprises several medical applications. The analysis of drugs in human specimens can be conducted for clinical or legal/forensic purposes. Clinical applications include the acute management of overdose and therapeutic monitoring of drug concentrations to achieve maximum efficacy while limiting the toxicity and side effects of medications. Forensic applications of toxicology include analysis of drugs to provide evidence for civil and criminal court cases, to investigate the cause of death, to deter the use of performance-enhancing drugs in athletic competitions, and to determine operator impairment related to traffic citations and vehicle accidents. Workplace drug testing assesses pre-employment drug abuse and on-the-job impairment. Given the number of therapeutic drugs, drugs of abuse, and environmental toxins, as well as the variety of diseases, signs, and symptoms associated with drug exposure and overdose, there is an array of laboratory testing strategies. For this reason, the discussion of toxicology in this chapter will be divided into 3 broad sections—therapeutic drug monitoring (TDM), drugs of abuse, and environmental toxins (Figure 6–1).

Therapeutic Drug Monitoring
TDM is the practice of measuring the concentration of a drug or its metabolite in order to optimize the dosing of that drug to an individual patient and/or to assess patient compliance with a dosing schedule. The goal of TDM is to improve drug efficacy—the likelihood of a therapeutic effect while avoiding or minimizing adverse effects. Table 6–1 lists some commonly monitored drugs. Patients do not require monitoring for most drugs. However, for a limited group of agents or for patients with certain conditions (for instance, limited renal function, pregnancy, newborn or geriatric age groups), TDM plays an essential role in establishing the appropriate therapeutic dosing regimen.
The goal of therapeutic drug monitoring is to increase the likelihood of a therapeutic effect and avoid or minimize adverse effects. Patients do not require monitoring for most drugs.
| Methotrexate |
Immunosuppressants
|
Antibiotics
|
Antiepileptics (first generation)
|
Antiepileptics (second generation)
|
Tricyclic antidepressants
|
| Lithium |
Cardiac agents
|
Pain management
|
Prior to the 1960s, drug dosing was entirely empirical. For certain agents, this trial-and-error approach gave wide variations in patient response and a significant incidence of toxicity. Since then, physicians have learned to optimize drug dosages and delivery while avoiding many of the drug’s adverse effects. This has been achieved through the development of sensitive and rapid laboratory assays and the establishment of therapeutic ranges for common medications.
TDM is performed to optimize the dose of a drug to an individual patient. Drugs with a narrow therapeutic index or margin of safety (the difference between the effective dose and the toxic dose) are potential candidates for therapeutic monitoring (Table 6–2). TDM is useful for drugs that display significant pharmacokinetic variability that may be caused by drug interactions, genetic variation in drug metabolism, nonlinear kinetics, physiologic conditions such as pregnancy and aging, and underlying diseases that alter the effective amount of drug delivered to or metabolized by the body. When patient compliance is in question, drug monitoring may be used to demonstrate the presence or absence of the prescribed agent. TDM requires a suitable laboratory assay and the establishment of a therapeutic reference range that correlates with efficacy and/or toxicity.
| The prescribed drug has a low margin of safety; that is, toxic blood drug concentrations or dosages are only slightly greater than therapeutic ones (a narrow therapeutic index) |
| Patient compliance with their prescribed drug regimen is uncertain |
| The drug does not act via irreversible inhibition (“hit and run” effect) |
| Symptoms of underlying disease are difficult to distinguish from drug toxicity |
| The treatment goal is not an objectively measured end point (such as blood pressure) |
The prescribed drug has significant pharmacokinetic variability as a result of:
|
TDM is performed by measuring the concentration of a drug and metabolite(s). Blood or serum/plasma is the usual sample for TDM, but in some cases, urine or oral fluid samples are used to evaluate patient compliance. The most common examples of urine and oral fluid sampling are monitoring of buprenorphine, methadone, and oxycodone for compliance. By using blood levels to guide drug therapy, a proportional relationship is assumed between the plasma/serum concentration, the concentration of drug at the organ cellular level, and pharmacologic effect. For practical reasons, only blood levels of the drug are measured, because tissue concentrations cannot be easily sampled or analyzed. This pharmacokinetic principle of homogeneity defines the timing of sampling for TDM, since the concentrations of drug in blood at the moment of sample collection must reflect a proportional and constant (steady-state) concentration at the end organ and be reflective of drug effects at the cellular level. Most TDM samples are collected as trough concentrations, the lowest level just prior to the next dose, or as peak concentrations, 30 to 60 minutes after the dose, when blood levels are most reflective of the tissue concentration and drug efficacy or toxicity.
Pharmacokinetics is the study of drug interaction (absorption, metabolism, and clearance) within the body. Drug behavior in the body can be described by the LADME mnemonic. The “L” stands for liberation or release of the drug from its dosage form. The “A” is absorption that describes the movement of drug from the administration site into circulation. Distribution is the “D” and describes the reversible movement of drug through the circulatory system and body tissues. Metabolism or “M” is the chemical conversion of drug to active and inactive compounds. Finally, the “E” indicates how the body eliminates the drug.
Drugs behave in the body based on their chemical characteristics at the molecular level. Drugs can be acidic, basic, neutral, or polar (Table 6–3). The charge and dissociation constant (pK) of the drug influences its absorption, distribution, and elimination characteristics. The dissociation constant of the drug also affects how the drug can be extracted from patient samples and analyzed in the laboratory.
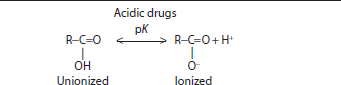 |
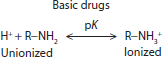 |
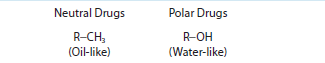 |
| The chemical groups on a drug determine the drug’s characteristics and behavior in the body |
| Acidic and basic chemical groups on drugs are in equilibrium between the unionized (uncharged) and ionized (charged) forms of the molecule |
| Unionized forms can passively diffuse while charged forms required active transport and bind to proteins and other counterions |
| Neutral drugs carry no charge and can be hydrophobic, oil-like, or polar, hydrophilic and attract water |
Drugs can be delivered to the body in a variety of ways. Patients may take a drug orally (PO) by pill (eg, aspirin) or dissolve a powder in a liquid drink (eg, laxatives). Drugs can be delivered intravascularly (IV) through a needle directly into circulation (antibiotics, like vancomycin). Some medications are delivered under the tongue, sublingual (SL), like nitroglycerin for cardiac pain or angina. Others may be injected under the skin, subcutaneous (SC), like compounds in a tuberculosis test, or intramuscularly (IM), such as vaccinations. Rectal (suppositories) and transdermal (eg, fentanyl pain patches) are other methods of delivering a drug. The route of administration will affect absorption and bioavailability of the drug to the body.
The route of drug administration and the formulation of the drug affect the rate and extent of drug absorption. For example, oral drug absorption is affected by many factors including drug solubility in enteral fluid, the acid–base characteristics of the drug, the lipid solubility of the drug, interferences with absorption by food, destruction of the drug by gastrointestinal flora, coadministration of other drugs—especially antacids, cholestyramine, and other resin-binding agents—blood flow to the gastrointestinal tract, and gastrointestinal transit time. Some orally delivered drugs are also subject to a significant “first-pass effect,” whereby they are largely metabolized by the liver to inactive compounds before reaching the systemic circulation. IV administration delivers drug directly into circulation bypassing “first-pass metabolism,” and the amount of drug delivered IV is often compared with other delivery options for determining the extent or amount of drug that is absorbed from a specific formulation. Significant variability in drug absorption is thus a common indication for TDM.
The chemical characteristics of a drug also affect the rate and extent of drug absorption. Acidic drugs carry a carboxyl group, R−COOH (Table 6–3). This acidic group is unionized or uncharged at pH levels below the drug’s dissociation constant and is ionized or charged (R−COO−) at pH levels above the drug’s dissociation constant. Drugs that act as strong acids have dissociation constants with a pK <5, such as salicylate, penicillin, and analgesics. Strongly acidic drugs are unionized and do not carry a charge at the acidic pH of the stomach while they carry a charge at the more basic pH of the intestines. So, drugs like salicylate are passively absorbed in the stomach, but require active transport for absorption across the intestines. Weak acids (barbiturates, sulfonamides, and thiazide diuretics) have dissociation constants in the range 5 to 11, and are preferentially absorbed in the intestines compared with the stomach. Strongly acidic drugs tend to be fully dissociated or charged at blood pH of 7.4 while weak acids may have significant amounts of the unionized form present in the blood. Due to the acidic pH of urine, weak acids that are ionized at blood pH may become unionized in urine and prone to greater reabsorption. Basic drugs contain an amine group (R−NH3). Basic groups are ionized (R−NH2+) below and unionized (uncharged) above their dissociation constant. Basic drugs can act as weak bases (eg, anesthetics, opiates, and antidepressants) with pK <10 or strong bases (eg, amphetamines and bronchodilators). Basic drugs tend to be significantly ionized (charged) at blood pH. Drugs can also be neutral and carry no charge across the range of physiologic pH. Neutral drugs can be lipophilic and act like fats (eg, corticosteroids) or polar and hydrophilic and attract water molecules (eg, digoxin).
After absorption, drugs distribute throughout the body through the circulatory system, lymphatic system, and tissue fluids. The amount of free drug available to act at organ receptors is affected by both protein and tissue binding. Protein binding is another consideration in TDM. Binding to plasma proteins occurs to some extent for most drugs, with bound and free (unbound) drugs existing in equilibrium. Although it is only the free drug fraction that is biologically active, most laboratory assays measure the total drug concentration, that is, the sum of the bound and the unbound drugs. Several factors can cause changes in plasma proteins and, consequently, affect free drug levels. Acid/neutral drugs tend to bind albumin while basic/neutral drugs bind α1-acid glycoprotein. Some drugs have specific binding proteins such as cortisol and corticosteroid-binding globulin, also known as transcortin. These proteins serve as transport proteins for drugs from the site of absorption to the tissue where drug can act, and as delivery mechanisms to the liver for metabolism or the kidney for elimination. Disease alterations in protein concentration can affect the concentration of free drug. For example, hypoalbuminemia, which occurs in the elderly and in patients with cirrhosis, may cause an increased free drug fraction in the setting of normal total drug levels. α1-Acid glycoprotein is an acute-phase reactant and levels of this protein increase in acute and chronic diseases. Increases in α1-acid glycoprotein create more binding sites for drug, so less free drug will be available in light of the same total drug concentration in a sample. The presence of uremia in disease results in compounds binding to albumin, displacing drug from the protein and elevating the free drug fraction. TDM can determine the proportion of free and total drugs in disease and individualize the dosage to the patient’s condition.
Drug metabolism typically renders nonpolar, lipophilic drugs into more polar, water-soluble compounds for elimination. The liver is the primary site for drug metabolism. Genetic variants, age, cirrhosis, and other hepatic conditions may adversely affect drug metabolism, and thus predispose a patient to toxicity. Many drugs are hepatic enzyme inducers or inhibitors and thus can influence the rate of their own metabolism, as well as the metabolism of many other drugs. Pharmacogenetics is study of drug action and metabolism based on genetics. Different genes can lead to changes in drug metabolism and produce an individualized response to a drug. Fast metabolizers will change parent drug to a metabolite at a greater rate than slow metabolizers, depending on the genes expressing metabolizing enzymes in the patient. For example, patients with the slow metabolizer gene will acetylate procainamide (a cardiac drug) to N-acetylprocainamide metabolite at a slower rate than fast metabolizers, and may be more prone to toxicity while on the same dose of drug.
Pharmacokinetics is the study of drug interaction (absorption, metabolism, and clearance) within the body. Pharmacogenetics is the study of drug metabolism based on genetics.
Elimination is the removal of drug and metabolites from the body. Drug can be eliminated through the kidneys, the liver, the lungs, the skin, the feces, and by other means. Elimination of many polar, nonlipophilic drugs is achieved primarily through renal excretion, which is dependent on adequate kidney function and renal blood flow. Other parameters relevant to elimination through the kidneys include urine pH and the properties of the drug itself, such as the dissociation constant, pK, and molecular size. Drug clearance is the theoretical volume of serum/plasma that is completely cleared of a drug per unit time. Importantly, clearance is the sum of all elimination mechanisms—hepatic, renal, lung, and any other—for a particular drug. Patients with impaired drug clearance may need more frequent monitoring.
In TDM, drug levels are most often determined only after steady state has been achieved. Steady state is the condition that occurs when the amount of drug entering the system equals the amount being eliminated. Steady-state concentration is compared in relation to a target range to determine changes in dosing. The target range is established from experimental dosing studies to determine the optimum drug concentration where a drug is most effective while causing the least undesirable side effects and toxicity. The target range is a generalized range that fits most patients, but that range may need to be adjusted or altered in certain disease states and physiologic conditions. TDM allows physicians to optimize drug dosage to a patient’s individual situation.
Most but not all drugs are eliminated by first-order (or linear) kinetics. This means that a constant fraction of total drug is eliminated per unit time. All drugs have a biological half-life. For drugs that follow first-order elimination kinetics, changes in dose will generally cause predictable changes in blood levels. Increases in drug concentration lead to increases in the rate of drug elimination. Some drugs, however, are eliminated by zero-order (or nonlinear) kinetics, such that a constant amount of drug is eliminated per unit time. Typically, metabolism by zero-order kinetics occurs when elimination pathways for that drug have been saturated. Under these circumstances, the biological half-life is not constant but depends on drug concentration. As a result, small increments in dose may cause disproportionately large increments in blood levels. Due to their lack of a predictable dose–response relationship, drugs that follow zero-order kinetics often require monitoring.
Most but not all drugs are eliminated by first-order (or linear) kinetics. This means that a constant fraction of drug is eliminated per unit time. Other drugs are eliminated by zero-order (or nonlinear) kinetics, such that a constant amount of drug is eliminated per unit time.
Assuming first-order kinetics, 5 half-lives are required after initiation of drug therapy to reach nearly complete (97%) steady state (5 half-life rule). Five half-lives are also required for nearly complete clearance of a drug after the termination of therapy, and for attaining a new steady state whenever a dosing regimen has been changed.
Many patients may take more than 1 medication, and those drugs can interact in the patient’s body. Drug interactions may cause displacement of bound drug from proteins. The clinical significance of the interaction is likely to be increased when both drugs are highly protein bound (80% or more), when 1 of the drugs has a higher binding affinity, or when 1 of the drugs is present in higher concentration than the other. Dosing adjustments may be required in these instances. Displacement of bound drug does not inevitably lead to an increased free drug level, because free drug is subject to increased metabolism and elimination. Increases in plasma proteins and drug binding may also occur as an acute-phase response or during pregnancy, and, consequently, higher dosing may be necessary. Caution must be used when interpreting total drug levels in patients with possible protein disturbances or drug interactions, and free drug levels may be more useful in these situations.
Currently, most clinical laboratories utilize immunoassays for the rapid and quantitative measurement of therapeutic drugs. In an immunoassay, drug in the patient’s sample competes with a drug conjugate (drug attached to an enzyme or fluorescein molecule) for the binding of specific antibodies. Antibody binding results in blocking enzyme activity or in enhancing fluorescence polarization. By measuring enzyme activity or fluorescence polarization, the amount of drug in the patient sample is quantitated. Chemiluminescent immunoassays offering superior sensitivity are also available for drug analysis. Other immunoassay methods such as ELISA and radioimmunoassay are less commonly used. More complex laboratory techniques, such as chromatography with ultraviolet or mass spectral detection, are also commonly utilized for drug measurements. (See Chapter 2.) Immunoassays offer advantages over chromatographic methods, because immunoassays can be automated and analyze a greater number of samples more rapidly with less labor and cost. Only the total drug concentrations are routinely measured. Free drug levels require a more time-consuming and expensive ultracentrifugation or dialysis equilibrium steps to separate the protein-bound drug from the free drug. Free drug concentrations are typically lower than total drug concentrations by a factor of 2- to 20-fold, so more sensitive assays are required.
The appropriate specimen for therapeutic drug measurements is usually serum or plasma. Most laboratories do not accept gel separator tubes as the gel can bind drugs and interfere with drug recovery. Immunosuppressant levels are measured using whole blood due to the distribution and concentration of drug in RBCs, which are removed in the preparation of serum/plasma. EDTA-anticoagulated whole blood is the appropriate sample for these immunosuppressant drug measurements. Urine samples are frequently used to evaluate patient compliance in cases of therapeutic administration of buprenorphine, methadone, and several opiates (including oxycodone). Saliva or oral fluid may be appropriate for monitoring some medications, such as theophylline, in pediatric patients or in those for whom phlebotomy is difficult. Oral fluid is also not subject to adulteration or substitution, which can be an issue with monitoring for pain management compliance in patients prone to abuse. In general, trough levels are drawn just prior to the next dose and are used to evaluate the likelihood of a therapeutic effect. Peak levels are drawn at varying times, depending on the particular drug, and are used typically to assess toxicity risk.
In general, trough levels are drawn just prior to the next dose and are used to evaluate the likelihood of a therapeutic effect. Peak levels are drawn at varying times, depending on the particular drug, and are used typically to assess toxicity risk.
Selected individual drugs and considerations for TDM are presented in Table 6–4. The required specimen volume and preservative will vary by analytical methodology, so the described collection instructions are only a guide. The reader should refer to specific instructions from the laboratory. The general monitoring recommendations will depend on the motivation for monitoring the drug, possible drug interactions, and whether the patient is stable or showing signs of toxicity. Therapeutic ranges are only suggestions and will vary by patient, condition, and the presence of other medications.
| Drug | Monitoring Recommendations | Specimen Collection Tube and Instructions | Suggested Therapeutic Range | Special Considerations |
|---|---|---|---|---|
| Methotrexate | 24, 48, 72 h after bolus; then daily until below cytotoxic levels | 5 mL red top; wrap in foil to protect from light; indicate time past bolus |
| Monitoring guidelines are for high-dose therapy (>20 mg/kg) only |
| Tacrolimus (FK-506) | Trough levels, 12 h post dose | 3 mL purple top | 5–20 ng/mL | Cross-reactivity with its metabolites in immunoassays |
| Cyclosporine | Trough levels, 12 or 24 h post dose | 3 mL purple top; avoid drawing from line of administration | Transplant of:
| Ranges depend on organ transplanted and time since transplant |
| Aminoglycosides | Peak:
| 5 mL red top |
| Guidelines for conventional dosing only (not low-dose therapy or pulse therapy) |
| Vancomycin | Either peak or trough, once per day | 5 mL red top | Peak: 30-40 μg/mL, trough: 5-10 μg/mL | Frequency of monitoring dependent on clinical situation |
| Phenytoin | Peak for toxicity is 4-5 h after dose; trough for monitoring | 5 mL red top | 10-20 μg/mL | Pertains to assay that measures total drug (free + bound) |
| Phenobarbital | Trough | 5 mL red top | 15-50 μg/mL | Steady state attained in 2-3 weeks |
| Carbamazepine | Peak level for toxicity is 2-4 h after dose; trough for monitoring | 5 mL red top | 4-12 μg/mL | Not helpful for idiosyncratic toxicities |
| Clonazepam | Peak for toxicity is 4 h after dose; trough for monitoring | 1 mL red or green top | 20-60 μg/mL | |
| Lamotrigine | Peak for toxicity is 2-4 h after dose; trough for monitoring | 1 mL red or green top | 3-14 μg/mL | |
| Levetiracetam | Peak for toxicity is 1 h after dose; trough for monitoring | 1 mL red or green top | 5-30 μg/mL | |
| Oxcarbazepine | Peak MHD for toxicity is 4-6 h after dose; trough for monitoring | 1 mL red or green top | 15-35 μg/mL MHD | |
| Valproic acid | Trough is not well defined | 5 mL red top | 50-100 μg/mL | Upper limit of therapeutic range |
| Tricyclic antidepressants | Steady state occurs in about 5 days; 10-14 h after once per day dosing; 4-6 h after twice per day dosing | 5 mL red top | Measure sum of parent and active metabolite for drugs noted with “a” in box at left | |
| Lithium | 10-14 h after dose; then biweekly or weekly until steady state; then every 1-3 months | 5 mL red top | 0.5-1.5 mmol/L (avoid green-top tubes) | Toxicity may occur at <1.5 mmol/L, especially in patients who show chronic toxicity |
| Digoxin | 8 h after PO dose; 12 h after IV dose; and at steady state (1 week after initiation) | 5 mL red top | 0.9-2.0 ng/mL | Specimen collection time is crucial to avoid falsely high levels; STAT levels occasionally necessary |
Methotrexate is a folate antagonist used in the treatment of a wide variety of neoplasms. Dose-related toxicity is common with high-dose methotrexate therapy (defined as >1 g/m2 or 20 mg/kg). Adverse effects include immunosuppression, and diverse organ damage including renal failure, myelosuppression, hepatic toxicity, neurotoxicity, gastrointestinal toxicity, and death. Toxicity correlates with serum methotrexate concentration and duration of exposure. Patients with poor hydration, renal insufficiency, pleural effusion, ascites, or gastrointestinal obstruction are at increased risk for toxicity. Adverse effects of methotrexate are ameliorated by administration of leucovorin, a reduced folate. Serial methotrexate levels are used to guide the appropriate dosing and duration of leucovorin rescue following high-dose methotrexate administration.
The immunosuppressant drugs, tacrolimus (FK-506), cyclosporin, and sirolimus (rapamycin), are drugs used to prevent rejection in organ transplantation. Cyclosporin is also utilized to treat psoriasis, chronic autoimmune urticaria, and rheumatoid arthritis. These drugs were originally discovered in bacteria (tacrolimus and sirolimus) and fungus (cyclosporin) from soil samples. Monitoring is indicated because these drugs have a narrow therapeutic index and highly variable pharmacokinetics. Adverse effects include nephrotoxicity, hepatotoxicity, pulmonary toxicity, neurotoxicity (light sensitivity, tingling in the palms, and tinnitus), tremor, and hypertension.
Whole blood is the preferred specimen for TDM, as the immunosuppressant drugs concentrate into erythrocytes more than the plasma/serum portion of blood. Low trough concentrations may indicate subtherapeutic immunosuppression and can be associated with increased risk of rejection. High trough concentrations cause increased toxicity including nephrotoxicity that can be particularly challenging to diagnose in renal transplant patients. Drug levels must be interpreted in conjunction with other laboratory test results and clinical findings to discriminate between toxicity and rejection. For renal transplant patients on cyclosporin therapy, the only definitive method for differentiating graft rejection from drug-induced nephrotoxicity is renal biopsy. These drugs are sometimes used in combination, and with mycophenolic acid, to enhance the immunosuppressant effects and decrease the dose and side effects.
The immunosuppressants are extensively metabolized by the liver to a number of metabolites, some of which have immunosuppressant activity. Some metabolites can cross-react in laboratory immunoassays, thus overestimating parent drug concentrations in situations where elimination is impaired and when metabolites accumulate, as in cholestasis. Patients who have received mouse monoclonal antibody therapies may also have inaccurate immunoassay results. HPLC with tandem mass spectrometry is increasingly being used for laboratory analysis to circumvent cross-reactivity with the immunoassays.
Gentamicin, tobramycin, and amikacin are aminoglycoside antibiotics. Ototoxicity and nephrotoxicity from aminoglycosides are related to dose and duration of exposure. Numerous factors, such as renal and cardiac function, age, liver disease, and obesity, affect the pharmacokinetic properties of aminoglycosides. Because of the many patient factors, as well as the low margin of safety and high incidence of dose-related toxicity, aminoglycoside levels are usually indicated in conjunction with renal function monitoring to minimize toxicity. In patients with normal renal function and without underlying disease, the indication for drug monitoring is less well defined.
Vancomycin is a tricyclic glycopeptide antibiotic with significant dose-related nephrotoxicity and ototoxicity. The practice of measuring vancomycin levels emerged from the guidelines for aminoglycoside monitoring. However, the necessity for vancomycin monitoring is controversial, because a good correlation between serum vancomycin levels and efficacy or toxicity has yet to be definitively demonstrated. Adult patients with normal renal function may not require routine monitoring. Indications for monitoring include impaired or changing renal function, concomitant use of nephrotoxic drugs, altered volume of distribution (as in a burn injury victim), prolonged vancomycin use, higher than usual doses, and use in neonates, children, pregnant women, and patients with malignancy.



