Learning Objectives
Learn the physiology and biochemistry of the relevant hormones and other important mediators.
Understand the laboratory tests used in the diagnosis of the more commonly encountered disorders.
Identify the clinical disorders associated with each of the endocrine glands and the role of specific laboratory tests in their diagnosis.
Introduction
This chapter on endocrine disorders is divided into separate discussions of each of the endocrine glands. Each section begins with an overview of the physiology and biochemistry of the relevant hormones. In addition, because of the large number of (often complex) laboratory tests in endocrinology, each section has a brief description of the laboratory tests most frequently used to diagnose the disorders in that disease group. Tests for which either serum or plasma is an acceptable specimen for analysis are noted as serum tests. Tests specifically requiring plasma are indicated by inclusion of the word “plasma” before the test name. As with all other chapters, each disorder is presented with a description of the disease and information useful in establishing a diagnosis. Figure 22–1 shows a general approach to the patient with endocrine disease.

Thyroid
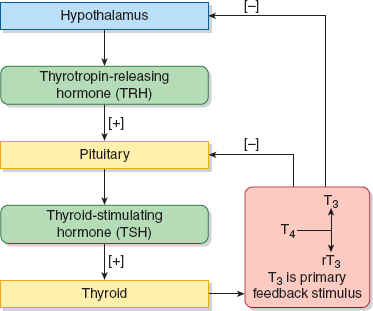
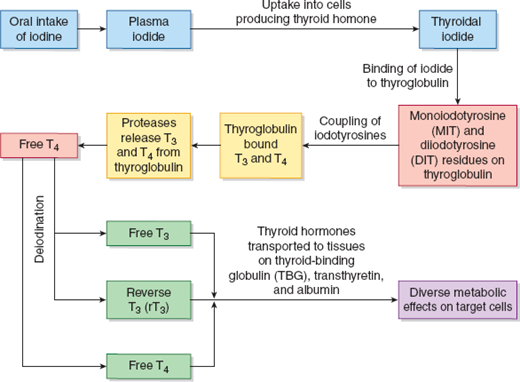
Production of thyroid hormones is regulated by the hypothalamic–pituitary–thyroid axis (Figure 22–2). Thyrotropin-releasing hormone (TRH) is produced in the hypothalamus and induces thyroid-stimulating hormone (TSH or thyrotropin) production in the anterior pituitary. TSH, in turn, stimulates thyroid hormone production and release by the thyroid gland. TSH production is inversely related to plasma thyroxine (T4) and triiodothyronine (T3) concentrations. The 2 primary hormones synthesized and secreted by the thyroid gland are T4 and, in lesser quantities, T3 (Figure 22–3). They are transported by plasma proteins—notably thyroid-binding globulin (TBG), transthyretin, and albumin—to various tissue sites where T4 is deiodinated to the active form, T3, and the inactive form known as reverse T3 (rT3). Thyroid hormones act through nuclear hormone receptors that are transcription factors for numerous genes. These genes regulate a number of critical physiologic functions in development and metabolism.
A “generational” classification has been applied for TSH assays based on the assay sensitivity. Third-generation assays can accurately measure TSH as low as 0.01 mU/L.
A “generational” classification has been applied for TSH immunoassays based on the assay sensitivity. Third-generation assays can accurately measure TSH as low as 0.01 mU/L. This allows the physician to distinguish mildly subnormal TSH values from the low values of overt hyperthyroid patients. The third-generation tests are also useful for evaluating the effectiveness of the thyroid hormone replacement in hypothyroid patients. Third-generation assays are essential for monitoring TSH suppression therapy in patients with a TSH-responsive thyroid tumor.
The relationship between TSH and the thyroid hormones, particularly free T4, is an inverse log-linear one, such that very small changes in free T4 result in large changes in TSH. Thus, TSH is the most sensitive first-line screening test for suspected thyroid abnormalities. If the TSH is within the normal reference range, no further testing is performed. If the TSH is outside of the reference range, a free T4 is obtained.
Assays are available for both total T4 and total T3 measurements. These assays are quite specific and suffer little interference. However, transient changes in serum thyroid hormone-binding protein concentrations may affect total T4 and T3 concentrations. Therefore, an assessment for free T4 (see below) is usually more helpful in evaluating thyroid function. The concentration of T4 in the blood is usually 100 to 200 times greater than the T3 level. In hyperthyroidism, total T3 and T4 concentrations correlate in all but a small subset of patients who have an elevation only in T3. For that reason, T3 should be measured in the serum of patients clinically suspected to be hyperthyroid and who have normal concentrations of serum T4. Measurement of T3 concentrations has limited clinical utility in assessment of hypothyroidism. Significant decreases in total T3 are seen in euthyroid sick syndrome (ETS).
“Direct” free thyroid hormone assays, without the need for a preliminary step to separate free hormones from hormones bound to protein carriers, are available for measurement of free T4 and free T3. Only a small fraction of T4 (<0.1%) circulates unbound to proteins, and this has made the accurate quantitation of free T4 analytically difficult. Free T4 is a better indicator of thyroid status than total T4 because, as noted above, the total T4 is altered by changes in the amounts of TBG, albumin, and other thyroid hormone-binding proteins. About 0.3% of T3 circulates as free T3. In general, free T3 concentrations correlate well with total T3, but as with T4, the concentrations of thyroid hormone-binding proteins influence the total T3 level.
Although the free T4 index offers an approximation of free T4, this measure is largely obsolete due to improvements in the free hormone assays. The free T4 index is calculated by multiplying the total T4 by the measure of available hormone-binding sites on TBG in an assay known as the percent T3 uptake. Because of the dependence on the amount of TBG, the free T4 index and the T3 uptake are affected by changes in the amount of thyroid-binding proteins induced by a variety of stimuli.
Antithyroid antibodies are present in approximately 15% of the general population and are the most common cause of thyroid disease in iodine-replete societies.
Under acute stress or in illness, there is a shift in the T4 deiodination in favor of the inactive rT3 form rather than the active T3. Numerous immunoassays are available for the measurement of rT3 using antisera that do not cross-react significantly with T3 or T4. rT3 is markedly increased in ETS syndrome (see below), but its measurement is rarely required for this diagnosis because its increase is proportional to the decrease in T3.
Antithyroid antibodies are present in approximately 15% of the general population and are the most common cause of thyroid disease in iodine-replete societies. They are also present in selected autoimmune diseases not usually associated with thyroid dysfunction. Descriptions of 3 types of antithyroid antibodies follow:
Antimicrosomal/antithyroid peroxidase antibodies (anti-TPO)—These antibodies are directed against a protein component of thyroid microsomes, the enzyme TPO. They are present in almost all patients with Hashimoto thyroiditis, in about 85% of patients with Graves disease (both discussed below), and in some patients with type 1 insulin-dependent diabetes mellitus, celiac disease, and Addison disease. An elevated titer of anti-TPO antibodies in the context of clinical symptoms of thyroid dysfunction and abnormal TSH and free T4 results is diagnostic for autoimmune thyroid disease. The presence of TPO antibodies before or during pregnancy is a good predictor of those women who will develop postpartum thyroid disease (discussed below). Normal concentrations are not well established because the antibodies may be found in healthy people (up to 12% of the population), and the reference range depends on the method used to perform the test.
Antithyroglobulin antibodies—These are also called colloidal antibodies. They are present in more than 85% of patients with Hashimoto thyroiditis and in more than 30% of patients with Graves disease. Like anti-TPO, antithyroglobulin antibodies also may be found in other autoimmune diseases. In iodine-sufficient areas, the antithyroglobulin antibody test is used less often, in favor of anti-TPO. However, in patients with suspected iodine deficiency, antithyroglobulin antibody is a better indicator of autoimmune thyroid disease.
TSH receptor antibodies—These are a diverse group of immunoglobulins that bind to TSH receptors and influence their action. They are found in most patients with Graves disease and in patients with selected other autoimmune disorders involving the thyroid. The biological functions of these antibodies vary from thyroid stimulation to thyroid inhibition (by blocking stimulation induced by TSH). Antibodies referred to as thyroid-stimulating immunoglobulins are present in 95% of patients with untreated Graves disease. In vitro bioassays can assess the ability of stimulatory antibodies to induce functional responses in cultured cells by measuring cyclic adenosine monophosphate increases or adenylate cyclase activity. Assays are available that measure the capability of the inhibitory antibodies, called thyrotropin-binding inhibitory immunoglobulins, to block the binding of labeled TSH to its receptors.
The radioactive iodine uptake and thyroid scan measurements involve the in vivo administration of radioactive iodine. Accumulated radioactivity in the thyroid is measured at intervals within 24 hours using a gamma scintillation counter. A nuclear imaging scan that examines the anatomic distribution of radioactive iodine uptake within the thyroid gland also may be obtained. Radioactive iodine uptake studies may be helpful when the diagnosis is in question and in differentiating between possible causes of hyperthyroidism.
Thyroglobulin is stored in the follicular colloid of the thyroid as a prohormone. Thyroglobulin measurements are used to monitor treatment in thyroid cancer. Persistent serum thyroglobulin after thyroidectomy is evidence of incomplete ablation or metastatic thyroid cancer. Thyroglobulin concentrations should always be assessed in the context of an antithyroglobulin antibody test because these autoantibodies can cause false-positive or negative thyroglobulin results.
Calcitonin is a polypeptide hormone produced by the thyroidal C cells, whose primary function is in regulating calcium homeostasis. It is elevated in patients with C-cell hyperplasia and medullary thyroid carcinoma (MTC). Calcitonin measurements are used to determine when to perform prophylactic thyroidectomy on patients with familial MTC. In addition, it is used to assess prognosis and monitor recurrence of MTC post thyroidectomy.
FNA is the procedure of choice to collect a specimen for microscopic review to distinguish benign from malignant thyroid nodules. The sensitivity of thyroid FNA for detection of thyroid cancer and other disorders varies from 70% to 97%. It depends greatly on the quality of the specimens and the experience of the cytopathologist. Thyroglobulin can also be measured in lymph node FNA aspirates to diagnose and monitor thyroid cancer.
In the presence of a clinical history and physical examination consistent with hyperthyroidism, a diagnosis of hyperthyroidism (but not necessarily its cause) can be established by the demonstration of a low TSH level and a high free T4.
Hyperthyroidism Overview and Associated Disorders
Hyperthyroidism, also known as thyrotoxicosis, is a collection of disorders associated with excess thyroid hormone (Table 22–1). There are 4 main causes of hyperthyroidism: 1) overstimulation of the thyroid (elevated TSH, human chorionic gonadotropin [hCG], and/or TSH receptor autoantibodies [TRAbs]); 2) genetic mutations leading to increased synthesis and secretion of thyroid hormone (germline, sporadic, or tumor induced); 3) release of excess hormone from the thyroid (inflammation, infection, injury); 4) extrathyroidal sources of thyroid hormone (ectopic thyroid tissue or exogenous hormone). Patients with hyperthyroidism demonstrate a spectrum of hypermetabolic features, including nervousness, palpitations, muscle weakness, increased appetite, diarrhea, heat intolerance, warm skin, weight loss, and perspiration. Affected patients also may have exophthalmos, emotional changes, menstrual changes, and a fine tremor of the hands. In the presence of a clinical history and physical examination consistent with hyperthyroidism, a diagnosis of hyperthyroidism (but not necessarily its cause) can be established by the demonstration of a low TSH level and a high free T4. In uncommon situations, only the total T3 level is elevated and the serum free T4 is normal (T3 thyrotoxicosis). To determine the etiology of the hyperthyroidism, additional testing is usually necessary. Graves disease, toxic multinodular goiter (TMNG), and toxic adenoma account for the vast majority (>95%) of cases of hyperthyroidism. It should be noted that diffuse or focal enlargement of the thyroid gland, also known as goiter, can be associated with hyperfunction, normal function, and hypofunction of the gland.
| Disorder | Laboratory Test Results Suggestive of Diagnosis in the Appropriate Clinical Setting |
|---|---|
| Hyperthyroidism | |
| Graves disease | TSH low; free T4 high; in some cases, T3 is elevated and free T4 is normal; TRAbs or TSI elevated |
| Toxic multinodular goiter | TSH low; free T4 and T3 normal or high; normal or increased radioactive iodine uptake; thyroid scan with multiple areas of increased uptake surrounded by suppressed uptake |
| Toxic adenoma | TSH low; free T4 and T3 normal or high; normal or increased radioactive iodine uptake; thyroid scan with focal increased uptake in tumor surrounded by suppressed uptake in nontumor tissue |
| Subacute thyroiditis | TSH low; free T4 and T3 high; increased; decreased radioactive iodine uptake |
| Painless thyroiditis | TSH low; free T4 and T3 high; erythrocyte sedimentation rate normal; decreased radioactive iodine uptake |
| Hypothyroidism | |
| Hashimoto thyroiditis | TSH high; T4 normal and then low, preceding a decline in T3; anti-TPO and/or antithyroglobulin antibody positive |
| Ablative hypothyroidism | TSH high; free T4 and T3 low following procedure that ablates thyroid |
| Infantile hypothyroidism | TSH high; free T4 low in a newborn or infant |
| Euthyroid sick syndrome | TSH normal to high; free T4 normal; T3 low; rT3 high; concentrations of TSH and thyroid hormones vary throughout disease course |
Thyroid storm is a relatively uncommon, but life-threatening manifestation of hyperthyroidism caused by excess circulation of thyroid hormones. Symptoms of thyroid storm are similar, but much more severe than traditional hyperthyroidism, including a markedly high fever of 105°F to 106°F, tachycardia, hypertension, and neurological and gastrointestinal abnormalities. Thyroid storm is precipitated by acute illnesses such as sepsis, diabetic ketoacidosis, and preeclampsia, as well as surgical or other diagnostic or therapeutic actions such as radioactive iodine use, anesthesia, excessive thyroid hormone ingestion, or thyroid palpation. Thyroid storm is associated with a high fatality rate if not identified early. The diagnosis is based on the presence of clinical signs and symptoms of severe hyperthyroidism in the context of a precipitating cause. In addition, marked elevations in free and total T4 are common in thyroid storm. Total T3 is unreliable in this setting because concomitant nonthyroidal illness (NTI) may cause T3 to decrease significantly.
Graves disease is a relatively common hyperthyroid disorder occurring more frequently in women. It is an autoimmune disease caused by TSH receptor autoantibodies that bind to and stimulate TSH receptors resulting in autonomous production of thyroid hormone.
Graves disease is a relatively common hyperthyroid disorder occurring more frequently in women. It has a familial predisposition. It is an autoimmune disease caused by TSH receptor antibodies (TRAbs) that bind to and stimulate TSH receptors resulting in autonomous production of thyroid hormone. While many patients have the classic signs and symptoms of thyrotoxicosis, in elderly patients with Graves disease, apathy, muscle weakness, and cardiovascular abnormalities occur more often than hypermetabolic symptoms.
Laboratory tests show undetectable TSH and increased free T4. In some cases, the T3 is elevated and the T4 is normal. The differential diagnosis includes TMNG, toxic adenoma, painless and subacute thyroiditis, ectopic thyroid tissue, and anxiety states (see below for descriptions). Detection of TRAbs along with the results from radioactive iodine uptake and nuclear thyroid scans are helpful in distinguishing among these possibilities. There is usually increased radioactive iodine uptake in Graves disease. The pattern on imaging is diffuse.
The cause of hyperthyroidism in patients with TMNG is an apparent functional autonomy of certain areas within the thyroid gland. The disorder is seen more commonly in elderly patients. The degree of hyperthyroidism is generally less severe than that found in Graves disease. Cardiovascular symptoms are prominent, such as arrhythmias, atrial fibrillation, or congestive heart failure, with weakness and wasting.
Laboratory tests usually show low or undetectable TSH and normal or elevated free T4 and T3 concentrations and no evidence of thyroid autoantibodies. Patients with TMNG will have normal to high radioactive iodine uptake, and the thyroid scan shows iodine localized to active nodules.
Thyroid adenomas that secrete thyroid hormones and cause hyperthyroidism are known as toxic adenomas. Thyroid hormone synthesis by a toxic adenoma is usually independent of TSH regulation, and it results in suppression of TSH secretion. These tumors can usually be distinguished from TMNG and Graves disease by a radioactive iodine uptake study and thyroid scan because there is localized uptake in the adenoma and little or no uptake in surrounding thyroid tissue.
Subacute thyroiditis is produced by a viral infection that alters thyroid function. This disease usually lasts for months, with thyroid function eventually returning to normal. The patient often has an associated upper respiratory infection, fever, and local pain mimicking a sore throat or an earache.
Patients in the early stage of this disease may have hyperthyroidism, with elevated T4 and T3 concentrations and a low TSH. Laboratory findings also often include a high erythrocyte sedimentation rate and little to no radioactive iodine uptake. If the disease progresses and the thyroid hormones are depleted, the patient develops hypothyroidism with low T3 and T4 concentrations and an elevated TSH.
Postpartum thyroid disease is a transient inflammatory process that has an onset of 1 to 6 months postpartum. Although the etiology is unclear, it is thought to be caused by a rebound in the immune system in response to the general state of immunosuppression that occurs during pregnancy. This disease can present as either hyperthyroidism or hypothyroidism. The typical disease course begins with a period of hyperthyroidism with elevated free T4, reduced TSH, and little to no radioactive iodine uptake for 3 to 6 months. This is followed by a 3- to 6-month period of hypothyroidism associated with reduced concentrations of free T4 and elevated TSH that completely resolves after 1 year. Approximately 20% of women with postpartum hypothyroidism develop permanent disease, requiring lifelong treatment and monitoring. The presence of anti-TPO antibodies prior to and during pregnancy is associated with an increased risk for postpartum thyroiditis.
When hypothyroidism occurs during development and in infancy, it results in a condition known as cretinism, which is marked by retardation of physical and intellectual growth. When hypothyroidism first appears in older children and adults, the collection of signs and symptoms is known as myxedema.
Painless thyroiditis may be induced by numerous drugs including lithium, interferon, and in a small portion of patients on amiodarone therapy. Further, some patients with chronic thyroiditis have a transient painless thyrotoxicosis, of unclear etiology.
Typically, free T4 and T3 concentrations are elevated with a low TSH. Patients have a markedly depressed radioactive iodine uptake. Painless thyroiditis can be distinguished clinically from subacute thyroiditis because an elevated erythrocyte sedimentation rate and local pain in the region of the thyroid are more consistent with subacute thyroiditis. A definitive diagnosis can be made by microscopic review of cells obtained by aspiration or biopsy. Thyroglobulin measurements can differentiate among patients with chronic thyroiditis from those with thyrotoxicosis caused by surreptitious thyroid hormone intake.
When hypothyroidism occurs during development and in infancy, it results in a condition known as cretinism, which is marked by retardation of physical and intellectual growth. In 95% of cases, hypothyroidism originates in the thyroid gland itself. If a patient has an increased serum TSH and a decreased free T4—together with appropriate clinical symptoms—a diagnosis of hypothyroidism is confirmed (Table 22–1). In asymptomatic patients, increased TSH, accompanied by a normal free T4, is known as subclinical hypothyroidism and may be indicative of early stages of primary hypothyroidism. High titers of anti-TPO antibodies suggest Hashimoto thyroiditis (see below) or postpartum thyroid dysfunction in a postpartum woman. While in the United States, autoimmunity is the main cause of hypothyroidism, iodine deficiency is the primary cause worldwide.
Hypothyroidism also may be a result of inadequate stimulation of the thyroid by TSH. This is known as secondary hypothyroidism. A subnormal free T4 with a decreased or inappropriately normal TSH is suggestive of secondary hypothyroidism from decreased TSH production or production of a biologically inactive form of TSH in the pituitary. It is usually accompanied by other pituitary hormone deficiencies, and it is much less common than primary hypothyroidism.
Clinical pictures of hypothyroidism differ, depending on the age. Congenital hypothyroidism is characterized by low production of thyroid hormones and can result in growth and intellectual delay if untreated. In the United States, all states screen for congenital hypothyroidism by testing for elevated TSH or a combination of elevated TSH and decreased free T4. Significant changes in thyroid function occur in the neonatal period and throughout childhood. Therefore, TSH and free T4 concentrations should be assessed using age-specific reference intervals. In particular, T4 is typically elevated in newborns. In adults, hypothyroidism can have an insidious onset, especially in the elderly. Symptoms are usually nonspecific in the early stage and then progress to more definitive characteristics of hypothyroidism with dry hair, dry skin, periorbital puffiness, dull expression, large tongue, and enlarged heart. If untreated, myxedema coma with respiratory failure may occur. Treatment involves hormone replacement.
Hashimoto thyroiditis is a common chronic inflammatory disease of the thyroid that accounts for as many as 90% of all cases of hypothyroidism in areas of iodine sufficiency. Autoimmune factors are thought to be the cause. Hashimoto thyroiditis is often associated with other autoimmune diseases such as Sjögren syndrome and pernicious anemia.
Patients with Hashimoto thyroiditis carry anti-TPO and antithyroglobulin antibodies. Firm thyroid enlargement and goiter is characteristic, but atrophy is also seen. Patients typically have an increased TSH and may have a normal free T4 and an elevated radioactive iodine uptake in the early stage of the disease. Over time, serum T4 declines first, followed by a decline in T3 as hypothyroid symptoms become predominant.
Hashimoto thyroiditis is a common chronic inflammatory disease of the thyroid that accounts for as many as 90% of all cases of hypothyroidism. Patients with Hashimoto thyroiditis carry anti-TPO and antithyroglobulin antibodies.
Postablative hypothyroidism is a relatively common cause of hypothyroidism in adults. Thyroid ablation occurs with total or subtotal thyroidectomy, or following treatment with radioactive iodine for hyperthyroidism.
A history of ablative therapy along with an elevated TSH and a low free T4 concentration indicates that ablation has produced a hypothyroid state.
Severe hypothyroidism in infancy is known as cretinism and, as previously noted, is characterized by irreversible mental retardation and growth impairment unless treated promptly. The appearance of symptoms depends on the severity of the disorder. However, even severe hypothyroidism is not usually apparent at birth. Early diagnosis and treatment with thyroid hormone prevents the manifestations of the disease. Elevated TSH and a low T4 in a newborn or young infant are indicative of infantile hypothyroidism.
Normal pregnancy is associated with a number of physiologic changes in thyroid function resulting in differences in “normal” laboratory values for thyroid function tests. The increase in estrogen stimulates hepatic synthesis of TBG, resulting in a net increase in total T3 and total T4 by about 1.5-fold. Significant homology exists between hCG, the pregnancy-associated glycoprotein hormone (see Chapter 20), and TSH. Because of this, hCG can directly stimulate the thyroid to produce thyroid hormone. Excess production of thyroid hormone signals a downregulation of TSH secretion. In the first trimester of pregnancy, increasing hCG concentrations are directly mirrored by decreasing TSH concentrations, which return to low normal in the second and third trimesters. Thus, TSH measurements in pregnancy should be considered in the context of gestational age and a reduced upper limit of normal. Many laboratories now report TSH with trimester-specific reference intervals.
Thyroid dysfunction during pregnancy can result in increased risks for the mother and fetus. Hypothyroidism during pregnancy is associated with an increased risk of miscarriage or preterm delivery and impaired neurological development in the fetus. Although controversial, it is recommended to screen all high-risk and symptomatic pregnant women for hypothyroidism by measuring TSH. Hyperthyroidism in pregnancy is associated with an increased risk of spontaneous abortion, preterm delivery, preeclampsia, and thyroid anomalies in the newborn. A subnormal TSH test result should be followed with free T4 testing. An elevated free T4 with the presence of autoantibodies confirms the diagnosis of hyperthyroidism in pregnancy. In mothers with confirmed thyroid disease, fetal thyroid function can be evaluated with ultrasound and amniotic fluid testing for TSH, free T4, and total T4. Normal reference intervals are instrument-specific for amniotic fluid thyroid function tests.
It is estimated that 40% of emergency department patients have euthyroid sick syndrome (ETS) at presentation. Stress, trauma, and illness can alter thyroid hormone production, transport, and metabolism, and thereby TSH levels, because of disruption of the normal feedback relationship between TSH and T3 and T4. This condition with altered thyroid hormone levels and no intrinsic disorder of the thyroid gland is called ETS or NTI, of which there are several variants.
The condition with altered thyroid hormone levels and no intrinsic disorder of the thyroid gland is called euthyroid sick syndrome (ETS) or nonthyroidal illness (NTI), of which there are several variants.
There is no consensus in the literature regarding the diagnosis and also therapy of ETS. The cause of ETS is different from patient to patient and is dependent on the history and any endocrinologic diagnosis. In moderately ill patients with ETS, serum T4 concentrations are within the reference range, while serum T3 is decreased and rT3 is increased. Serum TSH concentrations are typically normal to low (except for a transient increase that may occur during recovery). However, it is not recommended to measure TSH in hospitalized patients unless there is a strong suspicion for thyroid disease. In seriously ill patients, ETS presents with low-normal T4 and significantly reduced total T3 concentrations. Serum rT3 is increased because of slow thyroid hormone clearance and greater than normal conversion of T4 to rT3 rather than to T3. An elevated rT3 in the appropriate clinical setting, with appropriately suggestive laboratory test results, points to ETS syndrome.
Masses or “nodules” in the thyroid may be associated with normal function, hyperfunction, or hypofunction, and for that reason, they are considered apart from hyperthyroidism and hypothyroidism. In fact, some studies suggest that 25% to 40% of the population have thyroid nodules. Most solitary masses detected with physical examination are the dominant nodule in a multinodular goiter, a cyst, or an asymmetric enlargement of the gland. Benign thyroid adenomas account for most of the neoplastic nodules. The initial evaluation for a thyroid nodule is to measure TSH and perform a thyroid ultrasound. If the TSH is suppressed, a nuclear scan can be performed. Hyperfunctioning nodules appear as “hot nodules” by nuclear scan because they take up radioactive iodine while uptake is suppressed in the remainder of the gland. A nonfunctional (cold) nodule carries an increased risk of malignancy and should be followed with ultrasound-guided FNA or biopsy. The morphologic variants of thyroid cancer, diagnosed with histopathologic review of a biopsy specimen or aspirate (in order of frequency), are papillary carcinoma, follicular carcinoma, medullary thyroid carcinoma, and anaplastic carcinoma.
The diagnosis is established by histopathologic review of a specimen obtained with FNA or biopsy. The accuracy of the diagnosis is increased with the use of guided ultrasound examination for sample collection. Treatment and risk of recurrence of thyroid neoplasms are assessed by periodic measurement of tumor markers, including thyroglobulin for papillary and follicular carcinomas, and calcitonin for MTC.
Adrenal Cortex
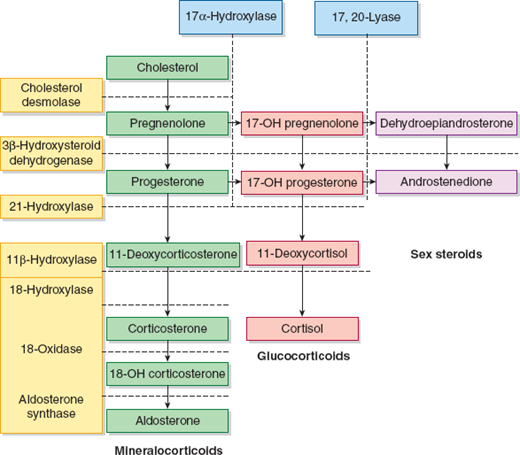
The adrenal cortex secretes many steroid hormones that have a wide variety of physiologic effects. The hormones can be grouped into 3 major categories: glucocorticoids, mineralocorticoids, and sex steroids that include androgens, progestogens, and estrogens. The glucocorticoids and mineralocorticoids are collectively known as corticosteroids. Steroid hormones are synthesized from cholesterol in the adrenal glands and in the gonads. They are transported in the blood bound to carrier proteins, such as albumin and hormone-binding globulins, or as free hormone. Steroids may be modified with glucuronate or sulfate to increase their water solubility and permit excretion via the kidneys or the gastrointestinal tract. The percentage of steroid hormone that is bound to protein varies with the hormone affinity for carrier proteins and ranges from 60% to nearly 100%. Quantitatively, the glucocorticoids and mineralocorticoids are the most important group of hormones produced by the adrenal cortex. The major corticosteroids are cortisol (a glucocorticoid) and aldosterone (a mineralocorticoid). The synthesis and metabolism of the steroid hormones are illustrated in Figure 22–4. The liver is the main site for conjugation of steroid hormones, and the kidney excretes approximately 90% of the conjugated steroids. Glucocorticoids alter carbohydrate metabolism by increasing gluconeogenesis and decreasing glucose utilization. Additional effects include the inhibition of amino acid uptake and protein synthesis in peripheral tissues. Mineralocorticoids promote sodium conservation and potassium loss and thereby considerably influence the retention or loss of fluid. Among the naturally occurring mineralocorticoids, aldosterone has the highest mineralocorticoid activity followed by deoxycorticosterone and corticosterone.
The adrenal cortex secretes hormones that can be grouped into 3 major categories: glucocorticoids, mineralocorticoids, and sex steroids that include androgens, progestogens, and estrogens.
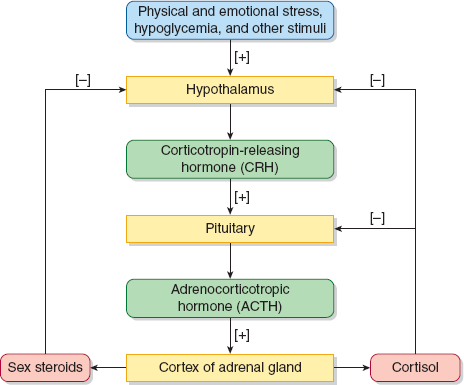
Secretion of adrenal glucocorticoids and adrenal androgens is regulated by corticotropin (ACTH) that is secreted by the pituitary gland (Figure 22–5). ACTH also plays a minor role in aldosterone and mineralocorticoid production. Their synthesis is primarily controlled by a different pathway known as the renin–angiotensin system. The hypothalamic–pituitary–adrenal (HPA) axis begins with the episodic release of corticotropin-releasing hormone (CRH) from the hypothalamus. CRH stimulates the episodic release of ACTH from the pituitary. ACTH then stimulates the adrenal cortex to produce cortisol in a diurnal or circadian manner. Cortisol concentrations are highest in the early morning between 4 and 8 AM and about 25% lower in the late evening. Physical and mental stress can elevate cortisol concentrations and blunt the circadian rhythm. ACTH release is under negative feedback control from the cortisol fraction not bound to proteins. Two adrenal sex steroids dehydroepiandrosterone (DHEA) and androstenedione are also stimulated by ACTH as well as other hormones including insulin-like growth factor-1 and gonadal steroids. Adrenal androgen production reaches a peak in the second decade, with a rise during late childhood. It gradually decreases and reaches a low level in the elderly.
Cortisol concentrations are highest in the early morning between 4 and 8 AM and about 25% lower in the late evening. Physical and mental stress can elevate cortisol concentrations and blunt the circadian rhythm.
As shown in Figure 22–6, aldosterone secretion is primarily controlled by the renin–angiotensin system. Renin is an enzyme synthesized and stored in cells of the juxtaglomerular afferent arterioles of the renal glomeruli. The circulating renin hydrolyzes angiotensinogen to produce angiotensin I, which is rapidly converted to angiotensin II by angiotensin-converting enzyme. Angiotensin II then stimulates the cells of the adrenal cortex to produce aldosterone. Angiotensin II is also a potent vasoconstrictor. The primary stimuli for renin release are decreased perfusion of the kidney and a negative sodium balance.
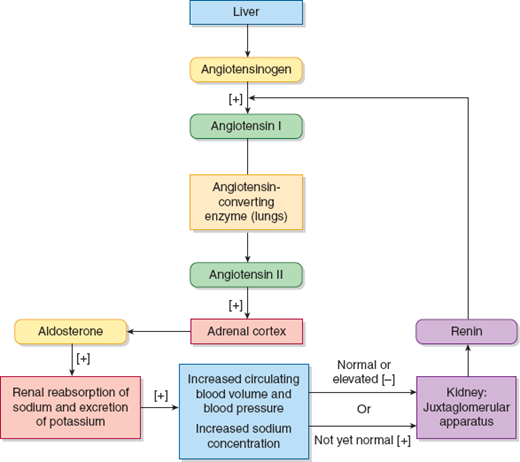
The functional status of the adrenal cortex can be evaluated by measuring the circulating concentrations of components of the HPA axis and the renin–angiotensin system. In addition to measurement of the plasma, serum, or urinary concentrations of these compounds, dynamic stimulation and suppression tests are valuable in identifying certain abnormalities.
Because the secretion of cortisol is diurnal and pulsatile, a single, random serum cortisol measurement is not useful in the diagnosis of adrenal dysfunction. However, a decreased early morning serum cortisol measurement may suggest adrenal insufficiency. The 24-hour urinary excretion of cortisol, an index of plasma-free cortisol during that 24-hour time frame, is a reliable gauge of excess cortisol secretion by the adrenal cortex. This is because taking an average urine cortisol concentration over 24 hours eliminates the need to account for diurnal variation as in a single serum collection. Urinary free cortisol (UFC) measurements should not be used in patients with renal impairment. Late-night salivary cortisol is also a measure of free cortisol since the binding protein does not cross into saliva. Salivary cortisol concentrations correlate well with serum concentrations and are not influenced by changes in cortisol-binding protein concentrations. Measurement of late-night salivary cortisol is an acceptable screening method for hypercortisolism.
Because the secretion of cortisol is pulsatile and diurnal, a single, random serum cortisol measurement is not usually diagnostic for adrenal dysfunction. The 24-hour urinary excretion of cortisol is a reliable gauge of excess cortisol secretion by the adrenal cortex.
Dexamethasone is a synthetic glucocorticoid more potent than cortisol that, when given orally or IV, suppresses ACTH and CRH secretion. It can be administered as 1 mg at midnight in an overnight dexamethasone suppression test, or in a 2-day low-dose dexamethasone suppression test (LDDST) by giving the patient 0.5 mg every 6 hours for 8 doses. If the dexamethasone is effective, it will suppress ACTH secretion and, thereby, suppress cortisol production. Patients with Cushing disease normally do not show suppression of cortisol synthesis after either dexamethasone administration for the LDDST or dexamethasone in the overnight suppression test.
In patients with an abnormal dexamethasone suppression test, ACTH measurements are important to determine whether Cushing syndrome is ACTH-dependent. The optimum time of day for determination of plasma ACTH concentration in patients with suspected Cushing syndrome is between midnight and 2 AM when the plasma-circulating concentrations of ACTH and cortisol are at their lowest point. At this time, the ability to detect an abnormality in ACTH secretion is the greatest. In patients with suspected adrenal insufficiency, ACTH should be measured in the morning during its peak. Suppressed morning ACTH may indicate excess adrenal cortisol secretion.
The ACTH stimulation test is the most useful test in diagnosis of adrenal insufficiency. It assesses the secretory response of the adrenal cortex to an ACTH-like stimulus. A 250-mg dose of an ACTH analog, Cortrosyn or cosyntropin, is administered IV or IM and serum cortisol is measured at 0, 30, and 60 minutes after injection. Normally, ACTH analogs will stimulate production of cortisol to concentrations >20 mg/dL. Lack of rise in cortisol after ACTH stimulation may indicate primary and secondary adrenal insufficiency.
CRH can be administered intravenously to determine the response of the pituitary to stimulation by hypothalamic hormone. The CRH stimulation test can be used to determine the source of adrenal insufficiency in patients with an abnormal ACTH stimulation test. Synthetic CRH is administered IV, and then ACTH and cortisol are measured at 0, 30, 60, 90, and 120 minutes after injection. Normal patients will respond to CRH by stimulating ACTH and cortisol production. Patients with primary adrenal insufficiency will have elevated ACTH, but no cortisol production, while patients with secondary disease will have both low ACTH and cortisol.
The most important test to establish a diagnosis of hyperaldosteronism or hypoaldosteronism is plasma aldosterone. Aldosterone can be measured in the plasma as the unmodified hormone, and in the urine as the 18-glucuronide conjugated metabolite of aldosterone. Screening for primary aldosteronism should include the determination of the ratio of plasma aldosterone concentration (PAC)/plasma renin activity (PRA), that is, the aldosterone to renin ratio (ARR). Aldosterone and renin concentrations are affected by numerous conditions and medication. Therefore, it is recommended that testing be performed under the following conditions: patients with normal potassium concentrations, patients with normal salt intake, patients removed from drugs that affect the ARR, and midmorning collection in patients who have been ambulatory for at least 2 hours.
PRA is assayed by measuring its ability to convert angiotensinogen to angiotensin I, which is then quantitated by an immunoassay. When primary aldosteronism is present, often because of either resistant or severe hypertension, the ARR will be elevated. Renin mass can also be assessed directly by immunoassay, to determine the direct renin concentration (DRC). Although the ARR can be calculated using DRC or PRA, fewer studies have investigated the utility of DRC. Renin specimens should not be stored refrigerated or on ice prior to analysis as cold temperatures promote secretion of prorenin, which falsely elevates results. See the section “Aldosterone” for recommended testing conditions.
Congenital adrenal hyperplasia (CAH) represents a spectrum of diseases resulting from enzyme deficiencies that impair normal hormone synthesis in the adrenal cortex. The decrease in cortisol production in most forms of CAH leads to an overproduction and shunting into the androgen synthesis pathway. These disorders are described in the section “Alterations in the Synthesis of Glucocorticoids, Mineralocorticoids, and Sex Steroids: Congenital Adrenal Hyperplasia.” The assays used to identify the specific enzyme deficiencies include measurement of 17-hydroxyprogesterone (17-OHP), either unstimulated or after ACTH stimulation, 17-hydroxypregnenolone after ACTH stimulation, deoxycorticosterone, 11-deoxycortisol, and several androgens (androstenedione, DHEA, and testosterone). DNA-based tests also can be used to identify certain gene mutations that result in enzyme deficiencies found in CAH.
Cushing syndrome is a disorder of excess cortisol production, which is more commonly ACTH-dependent than ACTH-independent.
Cushing syndrome is a disorder of excess cortisol production, which is more commonly ACTH-dependent than ACTH-independent. The early clinical features of Cushing syndrome are hypertension and weight gain. Truncal obesity with a round face, often known as a “moon facies,” and an accumulation of fat in the posterior neck and regions of the back close to the neck, known as a “buffalo hump,” appear with progression of the disease. Decreased muscle mass and proximal limb weakness occur from atrophy of muscle fibers induced by the high level of cortisol, which inhibits protein synthesis and uptake of glucose by the cells. Patients with Cushing syndrome often have elevated blood glucose levels with glucosuria. Other clinical signs and symptoms include striae of the skin, osteoporosis, hirsutism, menstrual abnormalities in women, and mental status changes involving mood swings with depression.
Cushing syndrome is separated into 3 main disease entities, with increased synthesis and loss of circadian rhythm resulting in excess cortisol production by the adrenal cortex as the common theme. Independent of the 3 naturally occurring forms of Cushing syndrome, the administration of glucocorticoids as a medication is a common cause of Cushing syndrome. This should be evident from the medication history. Endogenous forms of Cushing syndrome are described as follows:
Cushing disease is the most common form of Cushing syndrome and is 4- to 6-fold more prevalent in women. It is caused most often by small ACTH-secreting tumors in the pituitary (<1 cm in size) known as microadenomas. These adenomas can be detected with various radiographic techniques after appropriate hormone tests suggest a pituitary etiology. On rare occasions, the tumors are large and present as macroadenomas.
Adrenal Cushing syndrome—This form of Cushing syndrome is most commonly associated with a benign or malignant tumor in the adrenal cortex. Adrenal adenomas synthesize cortisol efficiently, but adrenal carcinomas often synthesize cortisol inefficiently and overproduce sex steroids, resulting in virilization.
Cushing syndrome from ectopic ACTH production—Small cell lung carcinoma and bronchial carcinoid patients account for most of the Cushing syndrome cases in this category. This form of Cushing syndrome is more common in men because of the higher incidence of lung cancer in men. In the absence of signs and symptoms specifically associated with the lung carcinoma or carcinoid, these patients are clinically indistinguishable from those with pituitary Cushing disease. Because the syndrome often appears in patients with significant clinical manifestations of cancer, the symptoms from ectopic ACTH production often go unrecognized. Another very rare cause of Cushing syndrome with an ectopic focus and high serum ACTH level is a tumor, most frequently a bronchial carcinoid tumor, which secretes CRH instead of ACTH.
The diagnosis of Cushing syndrome, or Cushing disease, discussed below collectively as Cushing syndrome, requires evidence of increased cortisol production and loss of suppression of cortisol synthesis or loss of cortisol diurnal variation. Screening for Cushing syndrome is difficult because 1) its prevalence is low; 2) several common conditions can produce biochemical and clinical signs of hypercortisolism in the absence of Cushing syndrome; and 3) the screening tests have a high rate of false-positive results for Cushing syndrome. For this reason it is recommended that a careful history be taken to exclude exogenous causes of hypercortisolism and that only patients with high clinical suspicion, such as those with unusual symptoms, for example, osteoporosis and less than 40 years old, children with a decrease in height and increase in weight percentiles, severe symptoms, or adrenal tumors visualized by imaging, be screened for Cushing syndrome.
The strategy for the diagnosis of Cushing syndrome, and the subsequent identification of 1 of the 3 forms of Cushing syndrome, involves tests to confirm endogenous hypercortisolism and then to determine whether the disease is ACTH-dependent. ACTH concentrations are low in patients with adrenal tumors, but normal or elevated with pituitary or ectopic ACTH-producing tumors. Table 22–2 summarizes the laboratory evaluation for Cushing syndrome.
The strategy for the diagnosis of Cushing syndrome, and the subsequent identification of 1 of the 3 forms of Cushing syndrome, involves tests to confirm endogenous hypercortisolism and then to determine whether the disease is ACTH-dependent.
| Laboratory Test | Pituitary Cause | Adrenal Cause | Ectopic ACTH Secretion |
|---|---|---|---|
| 24-Hour urine-free cortisol or late-night salivary cortisol | Elevated | Elevated | Elevated |
| Low-dose dexamethasone suppression test | No cortisol suppression | No cortisol suppression | No cortisol suppression |
| Plasma ACTH | Elevated or inappropriately normal | Low | For ectopic ACTH-secreting tumors, ACTH is elevated and often at higher levels than found in Cushing disease (pituitary cause) |
| Imaging study | MRI: pituitary tumor may be demonstrated | CT: adrenal tumor may be demonstrated | A tumor outside the adrenal may be demonstrated |
The following steps to diagnose and differentiate Cushing syndrome can be made using first-line and then second-line diagnostic tests, respectively.
It is recommended that at least one of the following testing strategies be performed to diagnose patients with Cushing syndrome:
The 24-hour UFC or late-night salivary cortisol is a sensitive screening test for patients with clinical signs and symptoms of Cushing syndrome. Because of the variability of the cortisol concentrations, the UFC or salivary cortisol should be elevated on at least 2 separate occasions in order to proceed with the diagnostic evaluation for Cushing syndrome. If the UFC or salivary cortisol is normal in a high-risk patient, an endocrinologist should be consulted for further studies. A low-risk patient with normal results should be rescreened in 6 months if signs or symptoms persist or worsen.
In unhealthy, but not critically ill, patients, and/or those with abnormal renal function, or disrupted sleep patterns, physicians should use one of the LDDSTs to screen for Cushing syndrome. If cortisol production is suppressed by a low dose of dexamethasone in an overnight or 48-hour test, Cushing syndrome is ruled out. If the clinical suspicion for Cushing syndrome is still high in the presence of a nonsuggestive result for Cushing syndrome, further evaluation by an endocrinologist may be useful. A lack of cortisol suppression after a low dose of dexamethasone is suggestive of Cushing syndrome. Patients taking oral estrogens should not be screened with the LDDST, as estrogens stimulate the liver to synthesize cortisol-binding globulin (CBG) and elevate serum cortisol concentrations, causing false-positive screening results. Further, critically ill patients synthesize less CGB and albumin, leading to lower serum cortisol concentrations, and possibly false-negative screening results.
Other clinical conditions that cause hypercortisolism should be ruled out. These include alcoholism, severe obesity, pregnancy, depression, diabetes mellitus, and glucocorticoid resistance.
Once Cushing syndrome is confirmed, a series of tests can then be performed to locate the cause of the hypercortisolism.
Plasma ACTH testing should be performed to determine the etiology of the hypercortisolism. If ACTH is suppressed, an adrenal source is suspected. If ACTH is high or inappropriately normal, the patient may have a pituitary adenoma.
If an ACTH-secreting pituitary adenoma is suspected, MRI of the pituitary gland to identify a mass or petrosal sinus blood sampling to collect samples coming directly out of the pituitary after CRH stimulation is used as a confirmation test. A ratio of petrosal ACTH to serum ACTH above suggested cutoff values before and after CRH administration is consistent with a pituitary cause.
If the ACTH is suppressed, a CT of the adrenal glands is likely to be informative to identify an adrenal tumor and indicate an adrenal cause.
If an ectopic ACTH-secreting tumor is suspected, this can be challenging to locate, but imaging studies are often valuable. Chest, pancreas, colon, and gall bladder carcinomas have been shown to be sources of cortisol secretion.
Adrenal insufficiency can be either primary, from destruction of the adrenal cortex by a local disease process or a systemic disorder, or secondary from pituitary or hypothalamic disease that reduces stimulation of the adrenal gland.
“Pseudo-Cushing syndrome,” which can be produced by alcohol abuse and other disorders, mimics both the clinical and biochemical features of the true syndrome.
Adrenal insufficiency can be either primary, from destruction of the adrenal cortex by a local disease process or a systemic disorder, or secondary from pituitary or hypothalamic disease that reduces stimulation of the adrenal gland. The most common causes of primary adrenal insufficiency, in which all classes of adrenal cortical steroids are deficient, are autoimmune adrenalitis and tuberculosis in endemic regions. In this particular disorder, the adrenal medulla and its catecholamine synthesis are spared. In other primary adrenal insufficiency disorders in which the adrenal gland is damaged, the adrenal medulla may be damaged along with the adrenal cortex. In secondary adrenal insufficiency, in which there is deficient stimulation of the adrenal gland because of pituitary or hypothalamic abnormalities, the adrenal medulla is not affected and aldosterone deficiency is not usually present. Aldosterone secretion is more dependent on angiotensin II stimulation of the adrenal cortex than on stimulation of the adrenal cortex by ACTH (as discussed below).
Primary adrenal insufficiency—There are many causes of primary adrenal insufficiency. Dysfunction in 1 or more sites in the HPA axis is the major cause of the primary adrenal insufficiency. Chronic primary adrenal insufficiency, also known as Addison disease, occurs mostly in adults. The most common causes are autoimmune disease (Western world) and tuberculous adrenalitis (worldwide). Other causes of primary adrenal insufficiency include fungal or viral infections (ie, histoplasmosis or HIV), and anatomic destruction of the adrenal glands through surgery, hemorrhage, or metastatic carcinomas. Primary adrenal insufficiency is characterized by hyperpigmentation of the skin and mucous membranes. The lack of negative feedback from adrenal cortisol leads to increased ACTH production by the pituitary. A degradation product of ACTH and its prohormone pro-opiomelanocortin is melanocyte-stimulating hormone (MSH). MSH stimulates melanin production and induces melanocyte hyperpigmentation. Hyperkalemia and hypotension may be present if there is deficient aldosterone (see the sections “Primary Hypoaldosteronism” and “Secondary Hypoaldosteronism”). Primary adrenal insufficiency usually has a gradual onset but may occur abruptly with a stressor such as critical illness or surgery. The rapid onset form of adrenal insufficiency is often the result of adrenal hemorrhage or thrombosis that impairs blood supply to the gland.
Acute primary adrenocortical failure (also called Addisonian crisis) can be triggered by a severe infection, sepsis, or abrupt withdrawal of steroids. It is a life-threatening emergency, characterized by abnormal electrolytes (critically high potassium and low sodium), hypotension, and hypoglycemia. Sudden death can occur if it is not treated promptly.
Secondary adrenal insufficiency—A deficiency of ACTH secretion from any cause can lead to adrenal insufficiency. Long-term glucocorticoid therapy can result in prolonged suppression of CRH from the hypothalamus and ACTH from the pituitary and transient adrenal insufficiency. Hypopituitarism as a result of postpartum hemorrhage (Sheehan syndrome), radiation, surgery, or injury may result in decreased ACTH production leading to reduced glucocorticoid synthesis.
Adrenal insufficiency may involve a deficiency of glucocorticoids or both glucocorticoids and mineralocorticoids. Depending on the extent of adrenal hormone deficiency, patients may have decreased serum cortisol alone or in combination with decreased plasma aldosterone.
The management of adrenal insufficiency first requires determination of the disease source (ie, primary or secondary), followed by an identification of the specific cause for the adrenal insufficiency. The tests that are useful in the diagnosis of primary and secondary adrenal insufficiency are shown in Table 22–3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


