KEY POINTS
*This chapter is dedicated to its previous author, Dr. Stephen Lowry, my mentor and friend.
Endogenous damage-associated molecular patterns (DAMPs) are produced following tissue and cellular injury. These molecules interact with immune and nonimmune cell receptors to initiate a “sterile” systemic inflammatory response following severe traumatic injury.
In many cases, DAMP molecules are sensed by pattern recognition receptors (PRRs), which are the same receptors that cells use to sense invading pathogens. This explains, in part, the similar clinical picture of systemic inflammation observed in injured and/or septic patients.
The central nervous system receives information with regard to injury-induced inflammation via soluble mediators as well as direct neural projections that transmit information to regulatory areas in the brain. The resulting neuroendocrine reflex plays an important modulatory role in the immune response.
Inflammatory signals activate key cellular stress responses (the oxidative stress response, the heat shock protein response, the unfolded protein response, autophagy, and programmed cell death), which serve to mobilize cellular defenses and resources in an attempt to restore homeostasis.
The cells, mediators, signaling mechanisms, and pathways that compose and regulate the systemic inflammatory response are closely networked and tightly regulated by transcriptional events as well as by epigenetic mechanisms, posttranslational modification, and microRNA synthesis.
Nutritional assessments, whether clinical or laboratory guided, and intervention should be considered at an early juncture in all surgical and critically ill patients.
Management of critically ill and injured patients is optimized with the use of evidence-based and algorithm-driven therapy.
OVERVIEW: INJURY-ASSOCIATED SYSTEMIC INFLAMMATORY RESPONSE
The inflammatory response to injury or infection occurs as a consequence of the local or systemic release of “pathogen-associated” or “damage-associated” molecules, which use similar signaling pathways to mobilize the necessary resources required for the restoration of homeostasis. Minor host insults result in a localized inflammatory response that is transient and in most cases beneficial. Major host insults, however, may lead to amplified reactions, resulting in systemic inflammation, remote organ damage, and multiple organ failure in as many as 30% of those who are severely injured. Recent data support this idea and suggest that severely injured patients who are destined to die from their injuries differ from survivors only in the degree and duration of their dysregulated acute inflammatory response.1,2
This topic is highly relevant because systemic inflammation is a central feature3 of both sepsis and severe trauma. Understanding the complex pathways that regulate local and systemic inflammation is necessary to develop therapies to intervene during overwhelming sepsis or after severe injury. Sepsis, defined by a systemic inflammatory response to infection, is a disease process with an incidence of over 900,000 cases per year. Further, trauma is the leading cause of mortality and morbidity for individuals under age 45.
In this chapter, we will review what is known about the soluble and cellular effectors of the injury-induced inflammatory response; how the signals are sensed, transduced, and modulated; and how their dysregulation is associated with immune suppression. We will also discuss how these events are monitored and regulated by the central nervous system. Finally, we will review how injury reprograms cellular metabolism, in an attempt to mobilize energy and structural stores to meet the challenge of restoring homeostasis.
THE DETECTION OF CELLULAR INJURY
Traumatic injury activates the innate immune system to produce a systemic inflammatory response in an attempt to limit damage and to restore homeostasis. It includes two general responses: (a) an acute proinflammatory response resulting from innate immune system recognition of ligands, and (b) an anti-inflammatory response that may serve to modulate the proinflammatory phase and direct a return to homeostasis (Fig. 2-1). This is accompanied by a suppression of adaptive immunity.4 Rather than occurring sequentially, recent data indicate that all three responses are simultaneously and rapidly induced following severe traumatic injury.2
Figure 2-1.
Schematic representation of the systemic inflammatory response syndrome (SIRS) after injury, followed by a period of convalescence mediated by the counterregulatory anti-inflammatory response syndrome (CARS). Severe inflammation may lead to acute multiple organ failure (MOF) and early death after injury (dark blue arrow). A lesser inflammatory response followed by excessive CARS may induce a prolonged immunosuppressed state that can also be deleterious to the host (light blue arrow). Normal recovery after injury requires a period of systemic inflammation followed by a return to homeostasis (red arrow). (Adapted with permission from Guirao X, Lowry SF. Biologic control of injury and inflammation: Much more than too little or too late. World J Surg. 1996;20:437. With kind permission from Springer Science + Business Media.)

The degree of the systemic inflammatory response following trauma is proportional to injury severity and is an independent predictor of subsequent organ dysfunction and resultant mortality. Recent work has provided insight into the mechanisms by which immune activation in this setting is triggered. The clinical features of the injury-mediated systemic inflammatory response, characterized by increased body temperature, heart rate, respirations, and white blood cellcount, are similar to those observed with infection (Table 2-1). While significant efforts have been devoted to establishing a microbial etiology for this response, it is now widely accepted that systemic inflammation following trauma is sterile. Although the mechanisms for the sterile response are less well understood, it is likely to result from endogenous molecules that are produced as a consequence of tissue damage or cellular stress, as may occur with hemorrhagic shock and resuscitation.5 Termed alarmins or damage-associated molecular patterns (DAMPs), these effectors, along with the pathogen-associated molecular patterns (PAMPs), interact with specific cell receptors that are located both on the cell surface and intracellularly.6 The best described of these receptors are members of the toll-like receptor family.
TERM | DEFINITION |
|---|---|
Infection | Identifiable source of microbial insult |
SIRS | Two or more of following criteria are met:
White blood cell count ≥12,000/μL or ≤4000/μL or ≥10% band forms |
Sepsis | Identifiable source of infection + SIRS |
Severe sepsis | Sepsis + organ dysfunction |
Septic shock | Sepsis + cardiovascular collapse (requiring vasopressor support) |
Trauma DAMPs are structurally diverse endogenous molecules that are immunologically active. Table 2-2 includes a partial list of DAMPs that are released either passively from necrotic/damaged cells or actively from physiologically “stressed” cells by upregulation or overexpression. Once they are outside the cell, DAMPs promote the activation of innate immune cells, as well as the recruitment and activation of antigen-presenting cells, which are engaged in host defense.7 The best-characterized DAMP with significant preclinical evidence for its release after trauma and with a direct link to the systemic inflammatory response is high-mobility group protein B1 (HMGB1). Additional evidence for the role of DAMP molecules in postinjury inflammation, including mitochondrial proteins and DNA, as well as extracellular matrix molecules, is also presented.
| DAMP MOLECULE | PUTATIVE RECEPTOR(S) |
|---|---|
HMGB1 | TLRs (2,4,9), RAGE |
Heat shock proteins | TLR2, TLR4, CD40, CD14 |
S100 protein | RAGE |
Mitochondrial DNA | TLR9 |
Hyaluronan | TLR2, TLR4, CD44 |
Biglycan | TLR2 and TLR4 |
Formyl peptides (mitochondrial) | Formyl peptide receptor 1 |
IL-1α | IL-1 receptor |
The best-characterized DAMP in the context of the injury-associated inflammatory response is HMGB1 protein, which is rapidly released into the circulation within 30 minutes following trauma. HMGB1 is highly evolutionarily conserved across species. It was first described as a constitutively expressed, nonhistone chromosomal protein that participated in a variety of nuclear events, including DNA repair and transcription. HMGB1 was also detected in the cytosol and extracellular fluids at low levels, although its function outside the cell was not clear. Subsequent studies have proven, however, that HMGB1 is actively secreted from immune-competent cells stimulated by PAMPs (e.g., endotoxin) or by inflammatory cytokines (e.g., tumor necrosis factor and interleukin-1). This process occurs outside the classic secretory pathway via a mechanism that is independent of endoplasmic reticulum and the Golgi complex. Moreover, recent data indicate that HMGB1 release can be regulated by the inflammasome.8 Stressed nonimmune cells such as endothelial cells and platelet also actively secrete HMGB1. Finally, passive release of HMGB1 can occur following cell death, whether it is programmed or uncontrolled (necrosis).
Once outside the cell, HMGB1 interacts with its putative receptors either alone or in concert with pathogenic molecules to activate the immune response, and in this way, functions as a proinflammatory cytokine. HMGB1 has been shown to signal via the toll-like receptors (TLR2, TLR4, TLR9), the receptor for advanced glycosylation end products (RAGE), CD24, and others. The activation of TLRs mainly occurs in myeloid cells, whereas RAGE is thought to be the receptor target in endothelial and somatic cells. The diverse proinflammatory biologic responses that result from HMGB1 signaling include: (a) the release of cytokines and chemokines from macrophages/monocytes and dendritic cells; (b) neutrophil activation and chemotaxis; (c) alterations in epithelial barrier function, including increased permeability; and (d) increased procoagulant activity on platelet surfaces, among others.9 In particular, HMGB1 binding to TLR4 triggers the proinflammatory cytokine release that mediates “sickness behavior.” This effect is dependent on the highly conserved domain structure of HMGB1 that can be recapitulated by a synthetic 20-amino acid peptide containing a critical cysteine residue at position 106.10
Recent data have explored the role of this cysteine residue, as well as two others that are highly conserved, in the biologic function of HMGB1. They demonstrate that the redox state of the three residues regulates the receptor binding ability of HMGB1 to influence its activity, including cytokine production. For example, a thiol at C106 is required for HMGB1 to promote macrophage tumor necrosis factor (TNF) release. In addition, a disulfide bond between C23 and C45 is also required for cytokine release because reduction of the disulfide linkage or further oxidation will reduce the ability of HMGB1 to function as a cytokine. Therefore, if all three cysteine residues are in reduced form, HMGB1 lacks the ability to bind and signal through TLR4, but gains the capacity to bind to CXCL12 to activate CXCR4 and serve as a chemotactic mediator. Importantly, shifts between the redox states have been demonstrated and indicate that redox state dynamics are important regulators of HMGB1.11
Importantly, HMGB1 levels in human subjects following injury correlate with the Injury Severity Score, complement activation, and an increase in circulating inflammatory mediators such as TNF.12 Unchecked, excessive HMGB1 has the capacity to promote a self-injurious innate immune response. In fact, exogenous administration of HMGB1 to normal animals produces fever, weight loss, epithelial barrier dysfunction, and even death.
Mitochondrial proteins and/or DNA can act as DAMPs by triggering an inflammatory response to necrosis and cellular stress. Specifically, the release of mitochondrial DNA (mtDNA) and formyl peptides from damaged or dysfunctional mitochondria has been implicated in activation of the macrophage inflammasome, a cytosolic signaling complex that responds to cellular stress. In support of this idea, plasma mtDNA has been shown to be thousands of times higher in both trauma patients and patients undergoing femoral fracture repair when compared to normal volunteers. Further, direct injection of mitochondria lysates in an animal model caused remote organ damage, including liver and lung inflammation.13 These data suggest that with stress or tissue injury, mtDNA and peptides are released from damaged mitochondria where they can contribute to a sterile inflammatory response. From an evolutionary perspective, given that eukaryotic mitochondria derive from bacterial origin, it would make sense that they retain bacterial features capable of eliciting a strong response that is typically associated with a pathogen trigger. For example, mtDNA is circular and contains hypomethylated CpG motifs that resemble bacterial CpG DNA. It is thus capable of producing formylated peptides, which potently induce an inflammatory phenotype in neutrophils, by increasing chemotaxis, oxidative burst, and cytokine secretion. In addition, the mitochondrial transcription factor A (TFAM), a highly abundant mitochondrial protein, is functionally and structurally homologous to HMGB1. It has also been shown be released in high amounts from damaged cells where it acts in conjunction with mtDNA to activate TLR9 signaling.14
Recent work has explored the role of extracellular matrix (ECM) proteins in the TLR-mediated inflammatory response that follows tissue injury. These molecules, which are sequestered under normal conditions, can be released in a soluble form with proteolytic digestion of the ECM. Proteoglycans, glycosaminoglycans, and glycoproteins such as fibronectin have all been implicated as key players in the DAMP/TLR interaction. Proteoglycans, in particular, have also been shown to activate the intracellular inflammasomes that trigger sterile inflammation. These molecules, which consist of a protein core with one or more covalently attached glycosaminoglycan chains, can be membrane-bound, secreted, or proteolytically cleaved and shed from the cell surface.
Biglycan is one of the first proteoglycans to be described as a TLR ligand.15 It consists of a protein core containing leucine-rich repeat regions, with two glycosaminoglycan (GAG) side chains (chondroitin sulfate or dermatan sulfate). Although biglycan typically exists in a matrix-bound form, with tissue injury, it is released from the ECM in a soluble form where it interacts with TLR2 or TLR4 to generate an immediate inflammatory response.
Various proinflammatory cytokines and chemokines, including TNF-α and interleukin (IL)-1β, are downstream effector molecules of biglycan/TLR2/4 signaling. Among these, the mechanism of biglycan-mediated autonomous synthesis and secretion of mature IL-1β is unique. Usually, release of mature IL-1β from the cell requires two signals, one which is needed to initiate synthesis (TLR2/4-mediated) and the other to process pro-IL-1β to its mature form (inflammasome-mediated). How is it possible for biglycan to provide both signals? Current evidence indicates that when soluble biglycan binds to the TLR, it simultaneously serves as a ligand for a purinergic receptor, which facilitates the inflammasome activation required for IL-1β processing.16 These data support the idea that DAMP-mediated signals can initiate a robust inflammatory response.
The inflammatory response that occurs following traumatic injury is similar to that observed with pathogen exposure. Not surprising, surface and cytoplasmic receptors that mediate the innate immune response to microbial infection have been implicated in the activation of sterile inflammation. In support of this idea, genes have been identified that are dysregulated acutely both in response to a microbial ligand administered to human volunteers and in response to traumatic injury in a large patient population.17 The classes of receptors that are important for sensing damaged cells and cell debris are part of the larger group of germline encoded pattern recognition receptors (PRRs). The best-described ligands for these receptors are microbial components, the PAMPs. The PRRs of the innate immune system fall into at least four distinct classes: TLRs, calcium-dependent (C-type) lectin receptors (CLRs), retinoic acid–inducible gene (RIG)-I-like receptors (RLRs), and the nucleotide-binding domain, leucine-rich repeat–containing (NBD-LRR) proteins (NLRs; also nucleotide-binding and oligomerization domain [NOD]-like receptors). Following receptor ligation, intracellular signaling modulates transcriptional and posttranslational events necessary for host defense by coordinating the synthesis and release of cytokines and chemokines to either initiate or suppress the inflammatory response. The best described of these, the TLRs, NLRs, and CLRs, are discussed in the following sections.
The TLRs are evolutionarily conserved type 1 transmembrane proteins that are the best-characterized PRRs in mammalian cells. They were first identified in Drosophila, where a mutation in the Toll gene led to its identification as a key component in their immune defense against fungal infection. The first human TLR, TLR4, was identified shortly thereafter. Now, more than 10 human TLR family members have been identified, with distinct ligands that include lipid, carbohydrate, peptide, and nucleic acid components of various pathogens. TLRs are expressed on both immune and nonimmune cells. At first, the expression of TLR was thought to be isolated to professional antigen-presenting cells such as dendritic cells and macrophages. However, mRNA for TLR family members have been detected in most cells of myeloid lineage, as well as natural killer (NK) cells.18 In addition, activation of T cells increases their TLR expression and induces their survival and clonal expansion. Direct engagement of TLR in T-regulatory (Treg) cells promotes their expansion and reprograms them to differentiate into T helper cells, which in turn provides help to effector cells. In addition, B cells express a distinct subset of the TLR family that determines their ability to respond to DAMPs; however, the significance of restricted TLR expression in these cells is not yet clear.
All TLRs consist of an extracellular domain, characterized by multiple leucine-rich repeats (LRRs), and a carboxy-terminal, intracellular toll/IL-1 receptor (TIR) domain. The LRR domains recognize bacterial and viral PAMPs in the extracellular environment (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) or in the endolysosomes (TLR3, TLR7, TLR8, TLR9, and TLR10). Although the role of TLRs in sepsis has been well described, more recent data indicate that a subset of the TLRs, TLR4 in particular, also recognizes DAMPs released from injured cells and tissues.19 Signal transduction occurs with receptor dimerization and recruitment of cytoplasmic adaptor proteins. These adaptor molecules initiate and amplify downstream signals, resulting in the activation of transcription. The transcription factors, which include nuclear factor-κB (NF-κB), activator protein (AP)-1, and interferon regulatory factor (IRF), bind to regulatory elements in promoters and/or enhancers of target genes leading to the upregulation of a large cohort of genes that include interferon (IFN)-α and IFN-β, nitric oxide synthase 2 (NOS2A), and TNF, which play critical roles in initiating innate immune responses to cellular injury and stress. Given the importance of TLR triggering of the innate immune response to immune homeostasis, it is no surprise that the process is tightly regulated. TLR expression is significantly increased following blunt traumatic injury. Further, TLR signaling is controlled at multiple levels, both posttranscriptionally via ubiquitination, phosphorylation, and microRNA actions that affect mRNA stability, as well as by the localization of the TLRs and their signaling complexes within the cell.
The NLRs are a large family of proteins composed of intracellular PRRs that sense both endogenous (DAMPs) and exogenous (PAMPs) molecules to trigger innate immune activation. The best characterized of the NLRs is the NLR family pyrin domain-containing 3 (NLRP3), which is highly expressed in peripheral blood leukocytes. It forms the key “sensing” component of the larger, multiprotein inflammasome complex, which is composed of NLRP3; the adapter protein apoptosis-associated speck-like protein containing a CARD (ASC); and the effector protein, caspase 1.20 In the cytoplasm, the receptor resides in an inactive form due to an internal interaction between two adjacent and highly conserved domains. In conjunction with a priming event, such as mitochondrial stress, phagocytosed DAMPs can be sensed by NLRP3, resulting in the removal of the self-repression. The protein can then oligomerize and recruit other complex members. The net result is the autoactivation of pro-caspase 1 to caspase 1. The NLRP3 inflammasome plays a central role in immune regulation by initiating the caspase 1–dependent processing and secretion of the proinflammatory cytokines IL-1β and IL-18. In fact, NLRP3 is the key protein in the mechanism by which IL-1β production is regulated in macrophages. NLRP3 inflammasome activity is tightly regulated by cell-cell interactions, cellular ion flux, and oxidative stress in order to maintain a balanced immune response to danger signals.
While the role of the NLRP3 inflammasome in the sterile inflammatory response following trauma has not been well described, recent evidence suggests that genetic variations in the NLRP3 gene might affect the magnitude of immune inflammatory responses following trauma. Single nucleotide polymorphisms within the NLRP3 gene were found to be associated with increased risk of sepsis and multiple organ dysfunction syndrome in patients with major trauma.21 In an animal model of burn injury, early inflammasome activation has been detected in a variety of immune cells (NK cells, CD4/CD8 T cells, and B cells), as determined by the assessment of caspase 1 cleavage by flow cytometry.22 Further, inhibition of caspase 1 activity in vivo results in increased burn mortality, suggesting that inflammasome activation may play an unanticipated protective role in the host response to injury that may be linked to increased production of specific cytokines. In addition to the NLRP3 inflammasome, there are numerous other NLRP sensors that are capable of detecting a diverse range of molecular targets. Among them are those endogenous molecules that are released as a consequence of tissue injury and cellular stress (hypoxia/hypoperfusion).
Macrophages and dendritic cells possess receptors that detect molecules released from damaged or dying cells in order to retrieve and process antigens from cell corpses for T-cell presentation. A key family of receptors that directs this process is the CLR family that includes the selectin and the mannose receptor families and that binds carbohydrates in a calcium-dependent fashion. Best described for their sensing of PAMPs, particularly fungal antigens, the CLRs can also act to promote the endocytosis and clearance of cell corpses. More recent work has demonstrated, however, that a subset of CLR receptors such as dendritic cell-NK lectin group receptor-1 (DNGR-1) and macrophage-inducible C-type lectin receptor (Mincle) recognize DAMPS of intracellular origin, such as F-actin and the ribonucleoprotein SAP-130.23 Ligation and activation of Mincle promotes its interaction with an Fcγ receptor, which contains immunoreceptor tyrosine-based activation motifs. This leads to proinflammatory cytokine, chemokine, and nitric oxide production, in addition to neutrophil recruitment. In this way, Mincle may contribute to local inflammation at sites of tissue injury.
Soluble pattern recognition molecules (PRMs) are a molecularly diverse group of molecules that share a conserved mode of action that is defined by complement activation, agglutination and neutralization, and opsonization. The best described of the PRMs are the pentraxins. PRMs can be synthesized at sites of injury and inflammation by macrophages and dendritic cells, while neutrophils can store PRMs and can release them rapidly following activation. In addition, epithelial tissues (the liver in particular) serve as a reservoir source for systemic mass release. The short pentraxin, C-reactive protein (CRP), was the first PRM to be identified. Serum amyloid protein (SAP), which has 51% sequence similarity to human CRP, also contains the pentraxin molecular signature. CRP and SAP plasma levels are low (≤3 mg/L) under normal circumstances. However, CRP is synthesized by the liver in response to IL-6, increasing serum levels more than a 1000-fold. Thus, CRP is considered part of the acute-phase protein response in humans. For this reason, CRP has been studied as a marker of the proinflammatory response in many clinical settings, including appendicitis, vasculitis, and ulcerative colitis. CRP and SAP are ancient immune molecules that share many functional properties with antibodies: they bind bacterial polysaccharides, ECM components, apoptotic cells, and nuclear materials, as well as all three classes of Fcγ receptors (FcγR). Both molecules also participate in the activation and regulation of complement pathways. In this way, short pentraxins can link immune cells to the complement system.24
Finally, significant data support a role for pentraxin 3 (PTX3), a long pentraxin family member, in the “sterile” inflammatory response associated with cellular stress. While CRP is produced solely in the liver, PTX3 is produced by various cells in peripheral tissues, including immune cells. PTX3 plasma concentrations increase rapidly in various inflammatory conditions, including sepsis. Further, in a recent prospective study of polytraumatized patients, serum PTX3 concentrations were highly elevated, peaking at 24 hours. In addition, PTX3 concentrations at admission were associated with injury severity, whereas higher PTX3 serum concentrations 24 hours after admission correlated with lower probability for survival.25
As noted earlier, members of the TLR family respond to endogenous molecules released from damaged or stressed cells. In animal models, activation of TLRs in the absence of bacterial pathogens correlates with the development of critical illness including “sterile inflammation.” What we know about TLR signaling events has largely been derived from the TLR-mediated response to bacterial pathogens. However, it is likely that the intracellular adaptors required for signal transmission by TLRs in response to exogenous ligands are conserved and used for “damage” sensing of endogenous (“self”) ligands as well. The intracellular domain structure of TLRs is highly conserved and is characterized by a cytoplasmic toll/IL-1R homology (TIR) domain. Binding of ligand to the receptor results in a receptor dimer, either a homodimer (e.g., TLR4/TLR4) or heterodimer (e.g., TLR2/TLR1), which recruits a number of adaptor proteins to the TIR domains, through TIR-TIR interaction.26 With one exception (TLR3), the universal adaptor protein central to the TLR signaling complex is myeloid differentiation factor 88 (MyD88), a member of the IL-1 receptor subfamily. MyD88 works through the recruitment of a second TIR-containing adaptor, MyD88 adaptor-like protein (Mal), in the context of TLR4 and TLR2 signaling, which serves as a bridge between MyD88 and activated TLRs to initiate signal transduction. It is interesting that Mal’s adaptor function requires cleavage of the carboxy-terminal portion of the protein by caspase 1, a key effector of the inflammasome.27 This finding suggests an important synergy between TLRs and NLRs that may potentiate TLR-mediated signaling. There are three other TIR domain-containing adaptor proteins that are also important to TLR-signaling events; these are TIR-domain-containing adapter-inducing INF-β (TRIF), TRIF-related adaptor molecule (TRAM), and sterile α- (SAM) and HEAT/armadillo (ARM) motif-containing protein (SARM). Two of these, TRIF and TRAM, are involved in the MyD88-independent signaling pathways, which are activated by TLR3 and TLR4.
Signaling through the MyD88-dependent pathway results in the activation of numerous cytoplasmic protein kinases including IL-1 receptor–associated kinases (IRAK-1 and IRAK-4), resulting in an interaction with TNF receptor–associated factor 6 (TRAF6). TRAF6, an E3 ubiquitin ligase, forms a complex with two other proteins, which together activate the complex that subsequently phosphorylates IκB kinase (IKK)-β and the MAP kinases (MAPKs). Ultimately, the phosphorylation of IκB by the IKK complex and NEMO (NF-κB essential modulator) leads to its degradation, which frees NF-κB and allows its translocation to the nucleus and the transcription of NF-κB target genes. Simultaneously, MAPK activation is critical for activation of the activator protein-1 (AP-1) transcription factor, and thus production of inflammatory cytokines. The MyD88-independent pathway acts through TRIF to activate NF-κB, similar to the MyD88-dependent pathway. However, TRIF can also recruit other signaling molecules to phosphorylate interferon-regulatory factor 3 (IRF3), which induces expression of type I IFN genes.26
As discussed earlier, activation and assembly of the inflammasome in response to DAMP sensing result in the cleavage of pro-caspase 1 into two products. This event is pivotal to all known inflammasome signaling pathways. The caspase 1 products assemble to form the IL-1 converting enzyme (ICE), which cleaves the IL-1 cytokines, IL-1β, IL-18, and IL-33. This final step is required for activation and secretion of the cytokines from the cell.20 IL-1β and IL-18 are potent proinflammatory cytokines that promote key immune responses that are essential to host defense. Thus, the synthesis, processing, and secretion of these cytokines are tightly regulated, as successful cytokine release requires a two-step process. The first signal, which is typically TLR-mediated, initiates the synthesis and storage of the inactive cytokine precursors in the cytoplasm. The second signal, which is inflammasome-mediated, initiates proteolytic cleavage of the procytokine, which is a requirement for its activation and secretion from the cell. Of further interest, evidence has demonstrated that both IL-1β and IL-18 lack a signal sequence, which is usually necessary for those proteins that are destined for cellular export. These signal peptides target proteins to the endoplasmic reticulum (ER) and to the Golgi complex, where they are packaged for secretion from the cell through the classical secretory pathway. More than 20 proteins in addition to IL-1β and IL-18 undergo unconventional protein secretion independent of the ER and Golgi complex.28 The list includes signaling molecules involved in inflammatory, cell survival, and repair responses, such as HMGB1, IL-1α, galectins 1 and 3, and FGF2. Currently, the mechanisms responsible for unconventional protein secretion are not understood; however, the process is also evident in yeast under conditions of cellular stress. It makes evolutionary sense that a mechanism for rapid secretion of stored proteins essential to the stress response is highly conserved.
CENTRAL NERVOUS SYSTEM REGULATION OF INFLAMMATION IN RESPONSE TO INJURY
The central nervous system (CNS) communicates with the body through ordered systems of sensory and motor neurons, which receive and integrate information to generate a coordinated response. Rather than being an immune-privileged organ, recent work indicates that the CNS receives information with regard to injury-induced inflammation both via soluble mediators as well as direct neural projections that transmit information to regulatory areas in the brain (Fig. 2-2). How does the CNS sense inflammation? DAMPs and inflammatory molecules convey stimulatory signals to the CNS via multiples routes. For example, soluble inflammatory signaling molecules from the periphery can reach neurons and glial cells directly through the fenestrated endothelium of the circumventricular organs (CVO) or via a leaky blood brain barrier in pathologic settings such as may occur following a traumatic brain injury.29 In addition, inflammatory stimuli can interact with receptors located on the brain endothelial cells to generate a variety of proinflammatory mediators (cytokines, chemokines, adhesion molecules, proteins of the complement system, and immune receptors) that directly impact the brain parenchyma. Not surprising, this response is countered by potent anti-inflammatory signaling, a portion of which is provided by the hypothalamic-pituitary-adrenal (HPA) axis and the release of systemic glucocorticoids. Inflammatory stimuli in the CNS result in behavioral changes, such as increased sleep, lethargy, reduced appetite, and the most common feature of infection, fever.
Figure 2-2.
Neural circuit relaying messages of localized injury to the brain (nucleus tractus solitarius). The brain follows with a hormone release (adrenocorticotropic hormone [ACTH], glucocorticoids) into the systemic circulation and by sympathetic response. The vagal response rapidly induces acetylcholine release directed at the site of injury to curtail the inflammatory response elicited by the activated immunocytes. This vagal response occurs in real time and is site specific. EPI = epinephrine; IL-1 = interleukin-1; NOREPI = norepinephrine; TNF = tumor necrosis factor. (Adapted and re-created with permission from Macmillan Publishers Ltd. Tracey KJ. The inflammatory reflex. Nature. 2002;420:853. Copyright © 2002.)

Information regarding peripheral inflammation and tissue damage can also be signaled to the brain via afferent neural fibers, particularly those of the vagus nerve.30 These afferent fibers can interconnect with neurons that project to the hypothalamus to modulate the HPA axis. In addition, afferent vagal nerve impulses modulate cells in the brain stem, at the dorsal motor nucleus of the vagus, from which efferent preganglionic parasympathetic impulses originate. Axons from these cells, which comprise the visceromotor component of the vagus nerve, form an “inflammatory reflex” that feeds back to the periphery to regulate inflammatory signaling events.31 Although the mechanisms by which cholinergic signals from the CNS regulate immune cells in the periphery are incompletely understood, recent evidence has provided some mechanistic insight. The first line of evidence to support this idea is the observation that vagal stimulation reduces proinflammatory cytokine production from the spleen in several experimental models systems.32 This effect is dependent on both the vagal efferent signals and, in part, splenic catecholaminergic nerve fibers that originate in the celiac plexus and that terminate in a T-cell–rich area of the spleen. Interestingly, these signals propagated by adrenergic nerves result in measurable increases in acetylcholine (ACh) levels in the spleen. In addition, the resident immune cells in the spleen require the expression of cholinergic receptors, specifically α7 nicotinic acetylcholine receptors (α7nAChR), for the suppression of cytokine synthesis.33 How is this effect mediated? The apparent source of ACh is choline-acetyltransferase–expressing T cells, which compose 2% to 3% of CD4+ T cells in the spleen and are capable of ACh production. Data also indicate that the vagus nerve may regulate inflammation in tissues that it directly innervates.
Traumatic injury results in complex neuroendocrine signaling from the brain that serves to enhance immune defense and rapidly mobilize substrates necessary to meet essential energy and structural needs. The two principle neuroendocrine pathways that orchestrate the host response are the hypothalamic-pituitary-adrenal (HPA) axis, which results in the release of glucocorticoid hormones, and the sympathetic nervous system, which results in release of the catecholamines, epinephrine, and norepinephrine. Virtually every hormone of the HPA axis influences the physiologic response to injury and stress (Table 2-3), but some with direct influence on the inflammatory response or immediate clinical impact are highlighted here, including growth hormone (GH), macrophage inhibitory factor (MIF), aldosterone, and insulin.
Hypothalamic Regulation Corticotropin-releasing hormone Thyrotropin-releasing hormone Growth hormone–releasing hormone Luteinizing hormone–releasing hormone Anterior Pituitary Regulation Adrenocorticotropic hormone Cortisol Thyroid-stimulating hormone Thyroxine Triiodothyronine Growth hormone Gonadotrophins Sex hormones Insulin-like growth factor Somatostatin Prolactin Endorphins Posterior Pituitary Regulation Vasopressin Oxytocin Autonomic System Norepinephrine Epinephrine Aldosterone Renin-Angiotensin System Insulin Glucagon Enkephalins |
One of the main mechanisms by which the brain responds to injury-associated stress is through activation of the HPA axis. Following injury, corticotrophin-releasing hormone (CRH) is secreted from the paraventricular nucleus (PVN) of the hypothalamus. This action is mediated in part by circulating cytokines produced as a result of the innate immune response to injury. These include TNF-α, IL-1β, IL-6, and the type I IFNs (IFN-α/β). Cytokines that are produced as a result of the adaptive immune response (IL-2 and IFN-γ) are also capable of increasing cortisol release. Direct neural input via afferent vagal fibers that interconnect with neurons projecting to the hypothalamus can also trigger CRH release. CRH acts on the anterior pituitary to stimulate the secretion of adrenocorticotropin hormone (ACTH) into the systemic circulation. Interestingly, the cytokines that act on the hypothalamus are also capable of stimulating ACTH release from the anterior pituitary so that marked elevations in ACTH and in cortisol can occur that are proportional in magnitude to the injury severity. Additionally, pain, anxiety, vasopressin, angiotensin II, cholecystokinin, vasoactive intestinal peptide, and catecholamines all contribute to ACTH release in the injured patient.
ACTH acts on the zona fasciculata of the adrenal glands to synthesize and secrete glucocorticoids (Fig. 2-3). Cortisol is the major glucocorticoid in humans and is essential for survival during significant physiologic stress. The resulting increase in cortisol levels following trauma have several important anti-inflammatory actions.

Cortisol elicits its many actions through a cytosolic receptor, the glucocorticoid receptor (GR). Because it is lipid soluble, cortisol can diffuse through the plasma membrane to interact with its receptor, which is sequestered in the cytoplasm in a complex with heat shock proteins (Fig. 2-4). Upon ligand binding, the GR is activated and can employ a number of mechanisms to modulate proinflammatory gene transcription and signaling events, with a “net” anti-inflammatory effect.34 For example, the activated GR complex can interact with transcription factors to sequester them in the cytoplasm, promote their degradation, or inhibit them through other mechanisms. Affected target genes include proinflammatory cytokines, growth factors, adhesion molecules, and nitric oxide. In addition, glucocorticoids can negatively affect the access of the transcription factor, NF-κB, to the promoter regions of its target genes via a mechanism that involves histone deacetylase 2. In this way, glucocorticoids can inhibit a major mechanism by which TLR ligation induces proinflammatory gene expression.35 The GR complex can also bind to specific nucleotide sequences (termed glucocorticoid response elements) to promote the transcription of genes that have anti-inflammatory functions. These include IL-10 and IL-1 receptor antagonist. Further, GR complex activation can indirectly influence TLR activity via an interaction with signaling pathways such as the mitogen-activated protein kinase and transforming growth factor–activated kinase-1 (TAK1) pathways. Finally, a recent report demonstrated that the GR complex can target both suppressor of cytokine signaling 1 (SOCS1) and type 1 IFNs to regulate TLR-induced STAT1 activation.36
Figure 2-4.
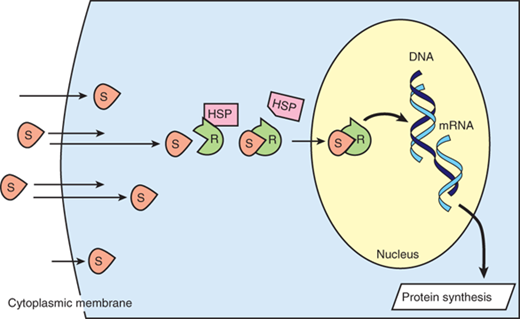
Simplified schematic of steroid transport into the nucleus. Steroid molecules (S) diffuse readily across cytoplasmic membranes. Intracellularly, the receptors (R) are rendered inactive by being coupled to heat shock protein (HSP). When S and R bind, HSP dissociates, and the S-R complex enters the nucleus, where the S-R complex induces DNA transcription, resulting in protein synthesis. mRNA = messenger RNA.
Adrenal insufficiency represents a clinical syndrome highlighted largely by inadequate amounts of circulating cortisol and aldosterone. Classically, adrenal insufficiency is described in patients with atrophic adrenal glands caused by exogenous steroid administration who undergo a stressor such as surgery. These patients subsequently manifest signs and symptoms such as tachycardia, hypotension, weakness, nausea, vomiting, and fever. Critical illness may be associated with a relative adrenal insufficiency such that the adrenal gland cannot mount an effective cortisol response to match the degree of injury. More recently, investigators have determined that critical illness-associated cortisol insufficiency in trauma patients occurs more frequently than previously thought.37 It has a bimodal presentation in which the patient is at increased risk both early following the injury-associated inflammatory response and in a delayed fashion, with sepsis being the initiating event. Laboratory findings in adrenal insufficiency include hypoglycemia from decreased gluconeogenesis, hyponatremia from impaired renal tubular sodium resorption, and hyperkalemia from diminished kaliuresis. Rigorous testing to establish the diagnosis includes monitoring of basal and ACTH-stimulated cortisol levels, both of which are lower than normal during adrenal insufficiency. Treatment strategies remain controversial; however, they include low-dose steroid supplementation.38
Macrophage inhibitory factor (MIF) is a proinflammatory cytokine expressed by a variety of cells and tissues, including the anterior pituitary, macrophages, and T lymphocytes. Several important functions of MIF in innate and adaptive immune responses and in inflammation have been described, supporting the idea that MIF may function to counteract the anti-inflammatory activity of glucocorticoids.39 For example, MIF has been reported to play a central role in the exacerbation of inflammation associated with acute lung injury, where it has been detected in the affected lungs and in alveolar macrophages. MIF has also been reported to upregulate the expression of TLR4 in macrophages.40 Finally, an early increase in plasma MIF has been detected in severely injured patients and was found to correlate with NF-κB translocation and respiratory burst in polymorphonuclear lymphocytes (PMNs) derived from severely injured patients. Further, nonsurvivors were shown to have higher serum MIF concentrations early after injury than survivors.41 These data suggest that targeting MIF after injury may be beneficial in preventing early PMN activation and subsequent organ failure in severely injured patients.
Growth hormone (GH) is a neurohormone expressed primarily by the pituitary gland that has both metabolic and immunomodulatory effects. GH promotes protein synthesis and insulin resistance and enhances the mobilization of fat stores. GH secretion is upregulated by hypothalamic GH-releasing hormone and downregulated by somatostatin. GH primarily exerts its downstream effects through direct interaction with GH receptors and through the enhanced hepatic synthesis of insulin-like growth factor (IGF)-1, an anabolic growth factor that is known to improve the metabolic rate, gut mucosal function, and protein loss after traumatic injury. Less than 5% of IGF-1 circulates free in the plasma, with the remainder bound principally to one of six IGF-binding proteins (IGFBPs), the majority to IGFBP-3. In the liver, IGF stimulates protein synthesis and glycogenesis; in adipose tissue, it increases glucose uptake and lipid utilization; and in skeletal muscles, it mediates glucose uptake and protein synthesis. In addition to its effects on cellular metabolism, GH enhances phagocytic activity of immunocytes through increased lysosomal superoxide production. It also increases the proliferation of T-cell populations.42 The catabolic state that follows severe injury has been linked to the suppression of the GH-IGF-IGFBP axis, as critical illness is associated with decreased circulating IGF levels. Not surprising, the administration of exogenous recombinant human GH (rhGH) has been studied in a prospective, randomized trial of critically ill patients where it was associated with increased mortality, prolonged ventilator dependence, and increased susceptibility to infection.43 More recently, circulating GH levels were examined on admission in 103 consecutive critically ill adult patients. In this study, circulating GH levels were about seven-fold increased in the 24 nonsurvivors when compared with survivors, and GH level was an independent predictor of mortality, along with the APACHE II/SAPS II scores. In distinct contrast, the effect of rhGH administration in severely burned children, both acutely and following prolonged treatment, has been proven to be beneficial. Pediatric burn patients receiving rhGH demonstrated markedly improved growth and lean body mass, whereas hypermetabolism was significantly attenuated.44 This finding was associated with significant increases in serum GH, IGF-1, and IGFBP-3.
Ghrelin, a natural ligand for the GH-secretagogue receptor 1a (GHS-R1a), is an appetite stimulant that is secreted by the stomach. GHS-R1a is expressed in a variety of tissues in different concentrations including the immune cells, B and T cells, and neutrophils. Ghrelin seems to play a role in promoting GH secretion and in glucose homeostasis, lipid metabolism, and immune function. In a rodent gut ischemia/reperfusion model, ghrelin administration inhibited proinflammatory cytokine release, reduced neutrophil infiltration, ameliorated intestinal barrier dysfunction, attenuated organ injury, and improved survival. It is interesting that this effect was dependent on an intact vagus nerve and that intracerebroventricular injection of ghrelin was also protective.45 These data suggest that the effect of ghrelin is mediated via the CNS, most likely through the “cholinergic anti-inflammatory pathway.” More recently, high ghrelin levels were demonstrated in critically ill patients as compared to healthy controls, independent of the presence of inflammatory markers. Moreover, the high ghrelin levels were a positive predictor of intensive care unit survival in septic patients, matching previous results from animal models.
Injury-induced activation of the sympathetic nervous system results in secretion of ACh from the preganglionic sympathetic fibers innervating the adrenal medulla. The adrenal medulla is a special case of autonomic innervation and is considered a modified postganglionic neuron. Thus, ACh signaling to the resident chromaffin cells ensures that a surge of epinephrine (EPI) and norepinephrine (NE) release into the circulation takes place in a ratio that is tightly regulated by both central and peripheral mechanisms. Circulating levels of EPI and NE are three- to four-fold elevated, an effect that persists for an extended time. The release of EPI can be modulated by transcriptional regulation of phenylethanolamine N-methyltransferase (PNMT), which catalyzes the last step of the catecholamine biosynthesis pathway methylating NE to form EPI. PNMT transcription, a key step in the regulation of EPI production, is activated in response to stress and tissue hypoxia by hypoxia-inducible factor 1α (HIF1A).
Catecholamine release almost immediately prepares the body for the “fight or flight” response with well-described effects on the cardiovascular and pulmonary systems and on metabolism. These include increased heart rate, myocardial contractility, conduction velocity, and blood pressure; the redirection of blood flow to skeletal muscle; increased cellular metabolism throughout the body; and mobilization of glucose from the liver via glycogenolysis, gluconeogenesis, lipolysis, and ketogenesis. To compound the resulting hyperglycemia, insulin release is decreased mainly through the stimulation of α-adrenergic pancreatic receptors. Hyperglycemia, as will be discussed later, contributes to the proinflammatory response and to further mitochondrial dysfunction.
The goal of this well-orchestrated catecholamine response is to re-establish and maintain the systems’ homeostasis, including the innate immune system. Circulating catecholamines can directly influence inflammatory cytokine production.46 Data indicate that basal EPI levels condition the activity and responsiveness of cytokine-secreting cells, which may explain large interindividual variability in innate cytokine profiles observed following injury. Epinephrine infusion at higher doses has been found to inhibit production of TNF-α in vivo and to enhance the production of the anti-inflammatory cytokine IL-10.47 Additionally, in vitro studies indicate that stress levels of glucocorticoids and EPI, acting in concert, can inhibit production of IL-12, a potent stimulator of Th1 responses. Further, they have been shown in vitro to decrease Th1 cytokine production and increase Th2 cytokine production to a significantly greater degree compared to either adrenal hormone alone. Thus, catecholamines secreted from the adrenal gland, specifically EPI, play a role in both innate proinflammatory cytokine regulation and adaptive Th responses, and may act in concert with cortisol during the injury response to modulate cytokine activity.48
How are these effects explained? It is well established that a variety of human immune cells (e.g., mononuclear cells, macrophages, granulocytes) express adrenergic receptors that are members of the family of G-protein–coupled receptors that act through the activation of intracellular second messengers such as cyclic adenosine monophosphate (cAMP) and calcium ion influx (discussed in more detail later). These second messengers can regulate a variety of immune cell functions, including the release of inflammatory cytokines and chemokines.
The sympathetic nervous system also has direct immune-modulatory properties via its innervation of lymphoid tissues that contain resting and activated immune cells. With stimulation of these postganglionic nerves, NE is released where it can interact with β2-adrenergic receptors expressed by CD4+ T and B lymphocytes, many of which also express α2-adrenergic receptors. Additionally, endogenous catecholamine expression has been detected in these cells, as has the machinery for catecholamine synthesis. For example, human peripheral blood mononuclear cells contain inducible mRNA for the catecholamine-generating enzymes, tyrosine-hydroxylase and dopamine-β-hydroxylase, and data suggest that cells can regulate their own catecholamine synthesis in response to extracellular cues. Exposure of peripheral blood mononuclear cells to NE triggers a distinct genetic profile that indicates a modulation of Th cell function. What the net effect of dopamine, NE, and EPI synthesis by circulating and resident immune cells may be relative to that secreted by the adrenal medulla is not clear and is an area that would certainly benefit from ongoing research efforts to identify novel therapeutic targets.
Aldosterone is a mineralocorticoid released by the zona glomerulosa of the adrenal cortex. It binds to the mineralocorticoid receptor (MR) of principal cells in the collecting duct of the kidney where it can stimulate expression of genes involved in sodium reabsorption and potassium excretion to regulate extracellular volume and blood pressure. MRs have also been shown to have effects on cell metabolism and immunity. For example, recent studies show aldosterone interferes with insulin signaling pathways and reduces expression of the insulin-sensitizing factors, adiponectin and peroxisome proliferator activated receptor-γ (PPAR-γ), which contribute to insulin resistance. In the immune system, mononuclear cells, such as monocytes and lymphocytes, have been shown to possess an MR that binds aldosterone with high specificity, regulating sodium and potassium flux, as well as plasminogen activator inhibitor-1 and p22 phox expression, in these cells.49 Further, aldosterone inhibits cytokine-mediated NF-κB activation in neutrophils, which also possess a functional MR.
Hyperglycemia and insulin resistance are hallmarks of injury and critical illness due to the catabolic effects of circulating mediators, including catecholamines, cortisol, glucagon, and GH. The increase in these circulating proglycemic factors, particularly EPI, induces glycogenolysis, lipolysis, and increased lactate production independent of available oxygen in a process that is termed “aerobic glycolysis.” Although there is an increase in insulin production at the same time, severe stress is frequently associated with insulin resistance, leading to decreased glucose uptake in the liver and the periphery contributing to acute hyperglycemia. Insulin is a hormone secreted by the pancreas, which mediates an overall host anabolic state through hepatic glycogenesis and glycolysis, peripheral glucose uptake, lipogenesis, and protein synthesis.50
The insulin receptor (IR) is widely expressed and consists of two isoforms, which can form homo- or heterodimers with insulin binding. Dimerization leads to receptor autophosphorylation and activation of intrinsic tyrosine kinase activity. Downstream signaling events are dependent on the recruitment of the adaptor proteins, insulin receptor substrate (IRS-1), and Shc to the IR. Systemic insulin resistance likely results from proinflammatory signals, which modulate the phosphorylation of IRS-1 to affect its function.
Hyperglycemia during critical illness is predictive of increased mortality in critically ill trauma patients.51 It can modulate the inflammatory response by altering leukocyte functions, and the resulting decreases in phagocytosis, chemotaxis, adhesion, and respiratory burst activities are associated with an increased risk for infection. In addition, glucose administration results in a rapid increase in NF-κB activation and proinflammatory cytokine production. Insulin therapy to manage hyperglycemia has grown in favor and has been shown to be associated with both decreased mortality and a reduction in infectious complications in select patient populations. However, the trend toward tight glycemic control in the intensive care unit failed to show benefit when examined in several reviews.52 Thus, the ideal blood glucose range within which to maintain critically ill patients and to avoid hypoglycemia has yet to be determined.
THE CELLULAR STRESS RESPONSES
Reactive oxygen and nitrogen species (ROS and RNS, respectively) are small molecules that are highly reactive due to the presence of unpaired outer orbit electrons. They can cause cellular injury to both host cells and invading pathogens through the oxidation of cell membrane substrates. Oxygen radicals are produced as a by-product of oxygen metabolism in the mitochondria as well as by processes mediated by cyclooxygenases, NADPH oxidase (NOX), and xanthine oxidase. The main areas of ROS production include mitochondrial respiratory chain, peroxisomal fatty acid metabolism, cytochrome P450 reactions, and the respiratory burst of phagocytic cells. In addition, protein folding in the endoplasmic reticulum can also result in the formation of ROS.53 Potent oxygen radicals include oxygen, superoxide, hydrogen peroxide, and hydroxyl radicals. RNS include NO and nitrite. The synthesis of ROS is regulated at several checkpoints and via several signaling mechanisms, including Ca2+ signaling, phosphorylation, and small G protein activation, which influence both the recruitment of the molecules required for NOX function and the synthesis of ROS in the mitochondria. NOX activation is triggered by a number of inflammatory mediators (e.g., TNF, chemokines, lysophospholipids, complement, and leukotrienes). Host cells are protected from the damaging effects of ROS through a number of mechanisms. The best described of these is via the upregulation and/or activation of endogenous antioxidant proteins. However, pyruvate kinase also provides negative feedback for ROS synthesis, as do molecules that react nonenzymatically with ROS. Under normal physiologic conditions, ROS production is balanced by these antioxidative strategies. In this context, ROS can act effectively as signaling molecules through their ability to modulate cysteine residues by oxidation and thus influence the functionality of target proteins.54 This has recently been described as a mechanism in the regulation of phosphatases. ROS can also contribute to transcription activity both indirectly, through its effects on transcription factor lifespan, and directly, through the oxidation of DNA. An important role for ROS has been well described in phagocytes, which use these small molecules for pathogen killing. Recent data, however, indicate that ROS may mediate inflammasome activation by diverse agonists.55 In addition, ROS appear to be involved in adaptive immunity. They have been described as a prime source of phosphatase activation in both B and T lymphocytes, which can regulate the function of key receptors and intracellular signaling molecules in these cells by affecting phosphorylation events.
Heat shock proteins (HSPs) are a group of intracellular proteins that are increasingly expressed during times of stress, such as burn injury, inflammation, oxidative stress, and infection. HSPs are expressed in the cytoplasm, nucleus, endoplasmic reticulum, and mitochondria, where they function as molecular chaperones that help monitor and maintain appropriate protein folding.56 HSPs accomplish this task through the promotion of protein refolding, the targeting of misfolded proteins for degradation, and the assistance of partially folded proteins to appropriate membrane compartments. HSPs bind also bind foreign proteins and thereby function as intracellular chaperones for ligands such as bacterial DNA and endotoxin. HSPs are presumed to protect cells from the effects of traumatic stress and, when released by damaged cells, alert the immune system of the tissue damage. However, depending on their location and the type of immune cell in which they are expressed, HSPs may exert proinflammatory immune activating signals or anti-inflammatory immune dampening signals (Table 2-4).57
| CELL LOCATION | RECOGNIZED AS DAMP? | IMMUNOMODULATORY FUNCTION | |
|---|---|---|---|
HSP90 | Cytoplasm, endoplasmic reticulum Can function both inside and outside the cell | May act as DAMP chaperone to activate innate immune response | Binds and optimizes RNA polymerase II action to regulate gene transcription Stabilizes glucocorticoid receptor in the cytoplasm Important for processing and membrane expression of TLR Chaperones include IKK Facilitates antigen presentation to dendritic cells |
HSP70 | Can function both inside and outside the cell Endoplasmic reticulum homolog is BiP | Exogenous HSP70 elicits cellular calcium flux, NF-κB activation, cytokine production | Can have anti-inflammatory actions when expression is increased Inhibits TLR-mediated cytokine production via NF-κB Reduces dendritic cell capacity for T-cell stimulation BiP sequesters proteins important to the unfolded protein response |
HSP60 | Mitochondria | Exogenous HSP60 inhibits NF-κB activation | Plays a role in intracellular protein trafficking Modulates cytokine synthesis |
Secreted, membrane-bound, and organelle-specific proteins fold in the lumen of the endoplasmic reticulum (ER) where they also receive their posttranslational modifications. Millimolar calcium concentrations are required to maintain the normal cellular protein folding capacity. Cellular stress decreases calcium concentration in the ER, disrupting the machinery required for this process and leading to the accumulation of misfolded or unfolded proteins. These occurrences are sensed by a highly conserved array of signaling proteins in the ER, including inositol requiring enzyme 1 (IRE1), protein kinase RNA (PKR)–like ER kinase (PERK), and activating transcription factor 6 (ATF6). Together, this complex generates the unfolded protein response (UPR), a mechanism by which ER distress signals are sent to the nucleus to modulate transcription in an attempt to restore homeostasis. Prolongation of the UPR, indicative of irreversible cellular damage, can result in cell death. Genes activated in the UPR result not only in the inhibition of translation, but also other potentially immunomodulatory events including induction of the acute-phase response, activation of NF-κB, and the generation of antibody-producing B cells.58
Burn injury leads to the marked reduction in ER calcium levels and activation of UPR sensing proteins. Moreover, recent data in a series of burn patients strongly link the UPR to insulin resistance and hyperglycemia in these patients.59 Thus, a better understanding of the UPR, which is triggered by severe inflammation, may allow the identification of novel therapeutic targets for injury-associated insulin resistance.
Under normal circumstances, cells need to have a way of disposing of damaged organelles and debris aggregates that are too large to be managed by proteasomal degradation. In order to accomplish this housekeeping task, cells use a process referred to as “macroautophagy” (autophagy), which is thought to have originated as a stress response.60 The steps of autophagy include the engulfment of cytoplasm/organelle by an “isolation membrane,” which is also called a phagophore. The edges of the phagophore then fuse to form the autophagosome, a double-membraned vesicle that sequesters the cytoplasmic material and that is a characteristic feature of autophagy. The autophagosome then fuses with a lysosome to form an autolysosome where the contents, together with the inner membrane, are degraded. This process is controlled by numerous autophagy-specific genes and by the specific kinase, mammalian target of rapamycin (mTOR).
As noted earlier, autophagy is a normal cellular process that occurs in quiescent cells for cellular maintenance. However, under conditions of hypoxia and low cellular energy, autophagy is induced in an attempt to provide additional nutrients for energy production. The induction of autophagy promotes a shift from aerobic respiration to glycolysis and allows cellular components of the autophagosome to be hydrolyzed to energy substrates. Increased levels of autophagy are typical in activated immune cells and are a mechanism for the disposal of ROS and phagocytosed debris.
Recent data support the idea that autophagy may also play an important role in the immune response.61 Autophagy is stimulated by Th1 cytokines and with activation of TLR in macrophages, but is inhibited by Th2 cytokines. It is also recognized as an important regulator of cytokine secretion, particularly those cytokines of the IL-1 family that are dependent on inflammasome processing for activation. For example, autophagosomes can sequester and degrade pro-IL-1β and inflammasome components. In animal models of sepsis, inhibition of autophagy results in increased proinflammatory cytokine levels that correlate with increased mortality.62 These data suggest that autophagy is a protective mechanism whereby the cell can regulate the levels of cytokine production.
Apoptosis (regulated cell death) is an energy-dependent, organized mechanism for clearing senescent or dysfunctional cells, including macrophages, neutrophils, and lymphocytes, without promoting an inflammatory response. This contrasts with cellular necrosis that results in disorganized intracellular molecule release with subsequent immune activation and inflammatory response. Systemic inflammation modulates apoptotic signaling in active immunocytes, which subsequently influences the inflammatory response through the loss of effector cells.
Apoptosis proceeds primarily through two pathways: the extrinsic pathway and the intrinsic pathway. The extrinsic pathway is activated through the binding of death receptors (e.g., Fas, TNFR), which leads to the recruitment of Fas-associated death domain protein and subsequent activation of caspase 3 (Fig. 2-5). On activation, caspases are the effectors of apoptotic signaling because they mediate the organized breakdown of nuclear DNA. The intrinsic pathway proceeds through protein mediators (e.g., Bcl-2, Bcl-2–associated death promoter, Bcl-2–associated X protein, Bim) that influence mitochondrial membrane permeability. Increased membrane permeability leads to the release of mitochondrial cytochrome C, which ultimately activates caspase 3 and thus induces apoptosis. These pathways do not function in a completely autonomous manner, because there is significant interaction and crosstalk between mediators of both extrinsic and intrinsic pathways. Apoptosis is modulated by several regulatory factors, including inhibitor of apoptosis proteins and regulatory caspases (e.g., caspases 1, 8, 10).
Figure 2-5.
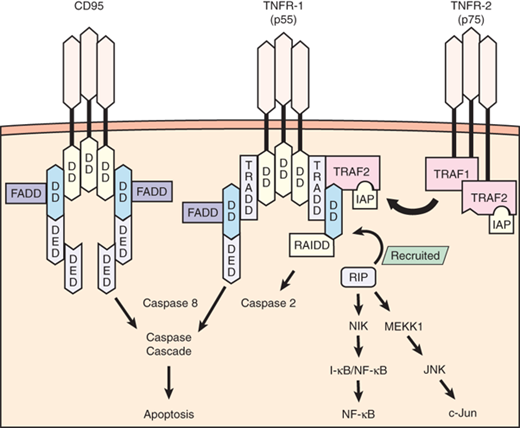
Signaling pathway for tumor necrosis factor receptor 1 (TNFR-1) (55 kDa) and TNFR-2 (75 kDa) occurs by the recruitment of several adapter proteins to the intracellular receptor complex. Optimal signaling activity requires receptor trimerization. TNFR-1 initially recruits TNFR-associated death domain (TRADD) and induces apoptosis through the actions of proteolytic enzymes known as caspases, a pathway shared by another receptor known as CD95 (Fas). CD95 and TNFR-1 possess similar intracellular sequences known as death domains (DDs), and both recruit the same adapter proteins known as Fas-associated death domains (FADDs) before activating caspase 8. TNFR-1 also induces apoptosis by activating caspase 2 through the recruitment of receptor-interacting protein (RIP). RIP also has a functional component that can initiate nuclear factor-κB (NF-κB) and c-Jun activation, both favoring cell survival and proinflammatory functions. TNFR-2 lacks a DD component but recruits adapter proteins known as TNFR-associated factors 1 and 2 (TRAF1, TRAF2) that interact with RIP to mediate NF-κB and c-Jun activation. TRAF2 also recruits additional proteins that are antiapoptotic, known as inhibitor of apoptosis proteins (IAPs). DED = death effector domain; I-κB = inhibitor of κB; I-κB/NF-κB = inactive complex of NF-κB that becomes activated when the I-κB portion is cleaved; JNK = c-Jun N-terminal kinase; MEKK1 = mitogen-activated protein/extracellular regulatory protein kinase kinase kinase-1; NIK = NF-κB–inducing kinase; RAIDD = RIP-associated interleukin-1b-converting enzyme and ced-homologue-1–like protein with death domain, which activates proapoptotic caspases. (Adapted with permission from Lin E, Calvano SE, Lowry SF. Tumor necrosis factor receptors in systemic inflammation. In: Vincent J-L (series ed), Marshall JC, Cohen J, eds. Update in Intensive Care and Emergency Medicine: Vol. 31: Immune Response in Critical Illness. Berlin: Springer-Verlag; 2002:365. With kind permission from Springer Science + Business Media.)
Apoptosis during sepsis may influence the ultimate competency of the acquired immune response. In a murine model of peritoneal sepsis, increased lymphocyte apoptosis was associated with mortality, which may be due to a resultant decrease in IFN-γ release. In postmortem analysis of patients who expired from overwhelming sepsis, there was an increase in lymphocyte apoptosis, whereas macrophage apoptosis did not appear to be affected. Clinical trials have observed an association between the degree of lymphopenia and disease severity in sepsis. In addition, after the phagocytosis of apoptotic cells by macrophages, anti-inflammatory mediators such as IL-10 are released that may exacerbate immune suppression during sepsis. Neutrophil apoptosis is inhibited by inflammatory products, including TNF, IL-1, IL-3, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IFN-γ. This retardation in regulated cell death may prolong and exacerbate secondary injury through neutrophil free radical release as the clearance of senescent cells is delayed.63
Cellular necrosis refers to the premature uncontrolled death of cells in living tissue typically caused by accidental exposure to external factors, such as ischemia, inflammation, or trauma, which result in extreme cellular stress. Necrosis is characterized by the loss of plasma membrane integrity and cellular collapse with extrusion of cytoplasmic contents, but the cell nuclei typically remain intact. Recent data have defined a process by which necrosis occurs through a series of well-described steps that are dependent on a signaling pathway that involves the receptor-interacting protein kinase (RIPK) complex. Termed “necroptosis,” it occurs in response to specific stimuli, such as TNF- and TLR-mediated signals.64 For example, ligation of the TNF receptor 1 (TNFR1) under conditions in which caspase 8 is inactivated (e.g., by pharmacologic agents) results in the overgeneration of ROS and a metabolic collapse. The net result is programmed necrosis (necroptosis). The effect of cell death by necroptosis on the immune response is not yet known. However, it is likely that the “DAMP” signature that occurs in response to necroptotic cell death is an important contributor to the systemic inflammatory response. Evidence to support this concept was provided by investigators who examined the role of necroptosis in murine models of sepsis. They demonstrated that Ripk3−/− mice were capable of recovering body temperature better, exhibited lower circulating DAMP levels, and survived at higher rates than their wild-type littermates.65 These data suggest that the cellular damage that occurs with programmed necrosis exacerbates the sepsis-associated systemic inflammatory response.
MEDIATORS OF INFLAMMATION
Cytokines are a class of protein signaling compounds that are essential for both innate and adaptive immune responses. Cytokines mediate a broad sequence of cellular responses, including cell migration, DNA replication, cell turnover, and immunocyte proliferation (Table 2-5). When functioning locally at the site of injury and infection, cytokines mediate the eradication of invading microorganisms and also promote wound healing. However, an exaggerated proinflammatory cytokine response to inflammatory stimuli may result in hemodynamic instability (i.e., septic shock) and metabolic derangements (i.e., muscle wasting). Anti-inflammatory cytokines also are released, at least in part, as an opposing influence to the proinflammatory cascade. These anti-inflammatory mediators may also result in immunocyte dysfunction and host immunosuppression. Cytokine signaling after an inflammatory stimulus can best be represented as a finely tuned balance of opposing influences and should not be oversimplified as a “black and white” proinflammatory/anti-inflammatory response. A brief discussion of the important cytokine molecules is included.
CYTOKINE | SOURCE | COMMENT |
|---|---|---|
TNF | Macrophages/monocytes Kupffer cells Neutrophils NK cells Astrocytes Endothelial cells T lymphocytes Adrenal cortical cells Adipocytes Keratinocytes Osteoblasts Mast cells Dendritic cells | Among earliest responders after injury; half-life <20 min; activates TNF receptors 1 and 2; induces significant shock and catabolism |
IL-1 | Macrophages/monocytes B and T lymphocytes NK cells Endothelial cells Epithelial cells Keratinocytes Fibroblasts Osteoblasts Dendritic cells Astrocytes Adrenal cortical cells Megakaryocytes Platelets Neutrophils Neuronal cells | Two forms (IL-1 α and IL-1 β); similar physiologic effects as TNF; induces fevers through prostaglandin activity in anterior hypothalamus; promotes β-endorphin release from pituitary; half-life <6 min |
IL-2 | T lymphocytes | Promotes lymphocyte proliferation, immunoglobulin production, gut barrier integrity; half-life <10 min; attenuated production after major blood loss leads to immunocompromise; regulates lymphocyte apoptosis |
IL-3 | T lymphocytes Macrophages Eosinophils Mast cells | |
IL-4 | T lymphocytes Mast cells Basophils Macrophages B lymphocytes Eosinophils Stromal cells | Induces B-lymphocyte production of IgG4 and IgE, mediators of allergic and anthelmintic response; downregulates TNF, IL-1, IL-6, IL-8 |
IL-5 | T lymphocytes Eosinophils Mast cells Basophils | Promotes eosinophil proliferation and airway inflammation |
IL-6 | Macrophages B lymphocytes Neutrophils Basophils Mast cells Fibroblasts Endothelial cells Astrocytes Synovial cells Adipocytes Osteoblasts Megakaryocytes Chromaffin cells Keratinocytes | Elicited by virtually all immunogenic cells; long half-life; circulating levels proportional to injury severity; prolongs activated neutrophil survival |
IL-8 | Macrophages/monocytes T lymphocytes Basophils Mast cells Epithelial cells Platelets | Chemoattractant for neutrophils, basophils, eosinophils, lymphocytes |
IL-10 | T lymphocytes B lymphocytes Macrophages Basophils Mast cells Keratinocytes | Prominent anti-inflammatory cytokine; reduces mortality in animal sepsis and ARDS models |
IL-12 | Macrophages/monocytes Neutrophils Keratinocytes Dendritic cells B lymphocytes | Promotes Th1 differentiation; synergistic activity with IL-2 |
IL-13 | T lymphocytes | Promotes B-lymphocyte function; structurally similar to IL-4; inhibits nitric oxide and endothelial activation |
IL-15 | Macrophages/monocytes Epithelial cells | Anti-inflammatory effect; promotes lymphocyte activation; promotes neutrophil phagocytosis in fungal infections |
IL-18 | Macrophages Kupffer cells Keratinocytes Adrenal cortical cells Osteoblasts | Similar to IL-12 in function; levels elevated in sepsis, particularly gram-positive infections; high levels found in cardiac deaths |
IFN-γ | T lymphocytes NK cells Macrophages | Mediates IL-12 and IL-18 function; half-life of days; found in wounds 5–7 d after injury; promotes ARDS |
GM-CSF | T lymphocytes Fibroblasts Endothelial cells Stromal cells | Promotes wound healing and inflammation through activation of leukocytes |
IL-21 | T lymphocytes | Preferentially secreted by Th2 cells; structurally similar to IL-2 and IL-15; activates NK cells, B and T lymphocytes; influences adaptive immunity |
HMGB1 | Monocytes/lymphocytes | High mobility group box chromosomal protein; DNA transcription factor; late (downstream) mediator of inflammation (ARDS, gut barrier disruption); induces “sickness behavior” |
TNF-α is a cytokine that is rapidly mobilized in response to stressors such as injury and infection and is a potent mediator of the subsequent inflammatory response. TNF is primarily synthesized by immune cells, such as macrophages, dendritic cells, and T lymphocytes, but nonimmune cells have also been reported to secrete low amounts of the cytokine.
TNF is generated in a precursor form called transmembrane TNF that is expressed as a trimer on the surface of activated cells. After being processed by the metalloproteinase TNF-α–converting enzyme (TACE; also known as ADAM-17), a smaller, soluble form of TNF is released, which mediates its biologic activities through type 1 and 2 TNF receptors (TNFR1; TNFR2).66 Transmembrane TNF-α also binds to TNFR1 and TNFR2, but its biologic activities are likely mediated through TNFR2. While the two receptors share homology in their ligand binding regions, there are distinct differences that regulate their biologic function. For example, TNFR1 is expressed by a wide variety of cells but is typically sequestered in the Golgi complex. Following appropriate cell signaling, TNFR1 is mobilized to the cell surface, where it sensitizes cells to TNF, or it can be cleaved from the surface in the form of a soluble receptor that can neutralize TNF.67 In contrast, TNFR2 expression is confined principally to immune cells where it resides in the plasma membrane. Both TNF receptors are capable of binding intracellular adaptor proteins that lead to activation of complex signaling processes and mediate the effects of TNF.
Although the circulating half-life of soluble TNF is brief, it acts upon almost every differentiated cell type, eliciting a wide range of important cellular responses. In particular, TNF elicits many metabolic and immunomodulatory activities. It stimulates muscle breakdown and cachexia through increased catabolism, insulin resistance, and redistribution of amino acids to hepatic circulation as fuel substrates. TNF also mediates coagulation activation, cell migration, and macrophage phagocytosis, and enhances the expression of adhesion molecules, prostaglandin E2, platelet-activating factor, glucocorticoids, and eicosanoids. Recent studies indicate that a significant early TNF response after trauma may be associated with improved survival in these patients.68
IL-1α and IL-1β, which are encoded by two distinct IL-1 genes, were the first described members of the IL-1 cytokine family. Currently, the family has expanded to 11 members, with the three major forms being IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1Rα). IL-1α and IL-1β share similar biologic functions, but have limited sequence homology. They use the same cell surface receptor, termed IL-1 receptor type 1 (IL-1R1), which is present on nearly all cells. Although IL-1Rα is synthesized and released in response to the same stimuli that lead to IL-1 production, it lacks the necessary domain to form a bioactive complex with the IL-1 receptor when bound. Thus, IL-1Rα serves as a competitive antagonist for the receptor. IL-1R activation initiates signaling events, which result in the synthesis and release of a variety of inflammatory mediators.
The IL-1α precursor is constitutively expressed and stored in a variety of healthy cells, including epithelium, endothelium, and platelets. Both the precursor and mature forms of IL-1α are active. With appropriate signals, IL-1α moves to the cell membrane where it can act on adjacent cells bearing the IL-1 receptor. It can also be released directly from injured cells. In this way, IL-1α is believed to function as a DAMP, which promotes the synthesis of inflammatory mediators, such as chemokines and eicosanoids. These mediators attract neutrophils to the injured site, facilitate their exit from the vasculature, and promote their activation. Once they have reached their target, neutrophil lifespan is extended by the presence of IL-1α.69
IL-1β, a multifunctional proinflammatory cytokine, is not detectable in healthy cells. Rather, its expression and synthesis occur in a more limited number of cells, such as monocytes, tissue macrophages, and dendritic cells, following their activation. IL-1β expression is tightly regulated at multiple levels (e.g., transcription, translation, and secretion), although the rate-limiting step is its transcription. IL-1β is synthesized and released in response to inflammatory stimuli, including cytokines (TNF, IL-18) and foreign pathogens. IL-1α or IL-1β itself can also induce IL-1β transcription. In contrast to IL-1α, IL-1β is synthesized as an inactive precursor molecule. The formation of mature IL-1β requires the assembly of the inflammasome complex by the cell and the activation of caspase 1, which is required for the processing of stored pro-IL-1β. Mature IL-1β is then released from the cell via an unconventional secretory pathway. IL-1β has a spectrum of proinflammatory effects that are largely similar to those induced by TNF, and injection of IL-1β alone is sufficient to induce inflammation. High doses of either IL-1β or TNF are associated with profound hemodynamic compromise. Interestingly, low doses of both IL-1β and TNF combined elicit hemodynamic events similar to those elicited by high doses of either mediator, which suggests a synergistic effect.
There are two primary receptor types for IL-1: IL-1R1 and IL-1R2. IL-1R1 is widely expressed and mediates inflammatory signaling on ligand binding. IL-1R2 is proteolytically cleaved from the membrane surface to soluble form on activation and thus serves as another mechanism for competition and regulation of IL-1 activity. IL-1α or IL-1β binds first to IL-1R1. This is followed by recruitment of a transmembrane coreceptor, termed the IL-1R accessory protein (IL-1RAcP). A complex is formed of IL-1R1 plus IL-1 plus the coreceptor. The signal is initiated with recruitment of the adaptor protein MyD88 to the toll–IL-1 receptor (TIR) domains of the receptor complex and signal transduction then occurs via intermediates, which are homologous to the signal cascade initiated by TLRs. These events culminate in the activation of NF-κB and its nuclear translocation.70
IL-2 is a multifunctional cytokine produced primarily by CD4+ T cells after antigen activation, which plays a pivotal role in the immune response. Other cellular sources for IL-2 include CD8+ and NK T cells, mast cells, and activated dendritic cells. Discovered as a T-cell growth factor, IL-2 also promotes CD8+ T-cell and NK cell cytolytic activity and modulates T-cell differentiation programs in response to antigen. Thus, IL-2 promotes naïve CD4+ T-cell differentiation into T helper 1 (Th1) and T helper 2 (Th2) cells while inhibiting T helper 17 (Th17) and T follicular helper (Tfh) cell differentiation. Moreover, IL-2 is essential for the development and maintenance of T regulatory (Treg) cells and for activation-induced cell death, thereby mediating tolerance and limiting inappropriate immune reactions. The upregulation of IL-2 requires calcium as well as protein kinase C signaling, which leads to the activation of transcription factors such as nuclear factor of activated T cells (NFAT) and NF-κB. MicroRNAs also play a role in the regulation of IL-2 expression.71
IL-2 binds to IL-2 receptors (IL-2R), which are expressed on leukocytes. IL-2Rs are formed from various combinations of three receptor subunits: IL-2Rα, IL-2Rβ, and IL-2Rγ; these form low-, medium-, and high-affinity forms of the receptor depending on the subunit combination. IL-2Rγ has been renamed the common cytokine receptor γ chain (γc), which is now known to be shared by IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Constitutive IL-2 receptor expression is low and is inducible by T-cell receptor ligation and cytokine stimulation. Importantly, the transcription of each receptor subunit is individually regulated via a complex process to effect tight control of surface expression. Once the receptor is ligated, the major IL-2 signaling pathways that are engaged include Janus kinase (JAK) signal transducer and activator of transcription (STAT), Shc-Ras-MAPK, and phosphoinositol-3-kinase (PI3K)-AKT. Partly due to its short half-life of <10 minutes, IL-2 is not readily detectable after acute injury. IL-2 receptor blockade induces immunosuppressive effects and can be pharmacologically used for organ transplantation. Attenuated IL-2 expression observed during major injury or blood transfusion may contribute to the relatively immunosuppressed state of the surgical patient.72
Following burn or traumatic injury, DAMPs from damaged or dying cells stimulate TLRs to produce IL-6, a pleiotropic cytokine that plays a central role in host defense. IL-6 levels in the circulation are detectable by 60 minutes, peak between 4 and 6 hours, and can persist for as long as 10 days. Further, plasma levels of IL-6 are proportional to the degree of injury. In the liver, IL-6 strongly induces a broad spectrum of acute-phase proteins such as CRP and fibrinogen, among others, whereas it reduces expression of albumin, cytochrome P450, and transferrin. In lymphocytes, IL-6 induces B-cell maturation into immunoglobulin-producing cells and regulates Th17/Treg balance. IL-6 modulates T-cell behavior by inducing the development of Th17 cells and inhibiting Treg cell differentiation in conjunction with transforming growth factor-β. IL-6 also promotes angiogenesis and increased vascular permeability, which are associated with local inflammatory responses. To date, 10 IL-6 family cytokines have been identified, including IL-6, oncostatin M, neuropoietin, IL-11, IL-27, and IL-31, all of which use trans signaling.73
The IL-6 receptor (IL-6R, gp80) is expressed on hepatocytes, monocytes, B cells, and neutrophils in humans. However, many other cells respond to IL-6 through a process known as trans signaling.74 In this case, soluble IL-6Rs (sIL-6R) exist in the serum and bind to IL-6, forming an IL-6/sIL-6R complex. The soluble receptor is produced by proteolytic cleavage from the surface of neutrophils in a process that is stimulated by CRP, complement factors, and leukotrienes. The IL-6/sIL-6R complex can then bind to the gp130 receptor, which is expressed ubiquitously on cells. Upon IL-6 stimulation, gp130 transduces two major signaling pathways: the JAK-STAT3 pathway and the SHP2-Gab-Ras-Erk-MAPK pathway, which is regulated by cytoplasmic suppressor of cytokine signaling (SOCS3). These signaling events can lead to increased expression of adhesion molecules as well as proinflammatory chemokines and cytokines. High plasma IL-6 levels have been associated with mortality during intra-abdominal sepsis.75
We have talked almost exclusively about the factors that initiate the inflammatory response following cellular stress or injury. The re-establishment of immune homeostasis following these events requires the resolution of inflammation and the initiation of tissue repair processes. IL-10 plays a central role in this anti-inflammatory response by regulating the duration and magnitude of inflammation in the host.
The IL-10 family currently has six members including IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26. IL-10 is produced by a variety of immune cells of both myeloid and lymphoid origin. Its synthesis is upregulated during times of stress and systemic inflammation; however, each cell type that produces IL-10 does so in response to different stimuli, allowing for tight control of its expression. IL-10 exerts effects by binding to the IL-10 receptor (IL-10R), which is a tetramer formed from two distinct subunits, IL-10R1 and IL-10R2. Specifically, IL-10 binds first to the IL-10R1 subunit, which then recruits IL-10R2, allowing the receptor complex to form. Whereas IL-10R2 is widely expressed, IL-10R1 expression is confined to leukocytes so that this differential expression of the receptor confines the effects of IL-10 to the immune system. Once receptor ligation occurs, signaling proceeds by the activation of JAK1 and STAT3. In particular, STAT3 in conjunction with IL-10 is absolutely required for the transcription of genes responsible for the anti-inflammatory response. IL-10 inhibits the secretion of proinflammatory cytokines, including TNF and IL-1, partly through the downregulation of NF-κB, and thereby functions as a negative feedback regulator of the inflammatory cascade.76 In macrophages, IL-10 suppresses the transcription of 20% of all lipopolysaccharide (LPS)-induced genes. Further, experimental models of inflammation have shown that neutralization of IL-10 increases TNF production and mortality, whereas restitution of circulating IL-10 reduces TNF levels and subsequent deleterious effects. Increased plasma levels of IL-10 also have been associated with mortality and disease severity after traumatic injury. IL-10 may significantly contribute to the underlying immunosuppressed state during sepsis through the inhibition and subsequent anergy of immunocytes. For example, IL-10 produced by Th2 cells directly suppresses Th1 cells and can feedback to suppress Th2 cell activity.77
IL-12 is unique among the cytokines in being the only heterodimeric cytokine. This family, which includes IL-12, IL-23, IL-27, and IL-35, consists of an α-chain that is structurally similar to the IL-6 cytokine and a β-chain that is similar to the class I receptor for cytokines. The individual IL-12 family members are formed from various combinations of the α and β subunits. Despite the sharing of individual subunits and the similarities of their receptors, the IL-12 cytokines have different biologic functions. IL-12 and IL-23 are considered proinflammatory, stimulatory cytokines with key roles in the development of Th1 and Th17 subsets of helper T cells. In contrast, both IL-27 and IL-35 appear to have immunoregulatory functions that are associated with cytokine inhibition in specific Treg cell populations, particularly the Th17 cells.78 The effects of these cytokines require specific receptor chains that are also shared among the cytokines. The complexity of signaling is evidenced by the fact that these receptor chains can function both as dimers and as monomers. Ligation of the IL-12 receptors initiates signaling events mediated by the JAK-STAT pathway. IL-12 synthesis and release are increased during endotoxemia and sepsis.79 IL-12 stimulates lymphocytes to increase secretion of IFN-γ with the costimulus of IL-18 and also stimulates NK cell cytotoxicity and helper T-cell differentiation in this setting. IL-12 release is inhibited by IL-10. IL-12 deficiency inhibits phagocytosis in neutrophils. In experimental models of inflammatory stress, IL-12 neutralization conferred a mortality benefit in mice during endotoxemia. However, in a cecal ligation and puncture model of intraperitoneal sepsis, IL-12 blockade was associated with increased mortality. Furthermore, later studies of intraperitoneal sepsis observed no difference in mortality with IL-12 administration; however, IL-12 knockout mice exhibited increased bacterial counts and inflammatory cytokine release, which suggests that IL-12 may contribute to an antibacterial response. IL-12 administration in chimpanzees is capable of stimulating the release of proinflammatory mediators such as IFN-γ and also anti-inflammatory mediators, including IL-10, soluble TNFR, and IL-1 receptor antagonists. In addition, IL-12 enhances coagulation as well as fibrinolysis.
IL-18 is a member of the IL-1 superfamily of cytokines. First noted as an IFN-γ–inducing factor produced by LPS-stimulated macrophages, IL-18 expression is found both in immune cells and nonimmune cells at low to intermediate levels. However, activated macrophages and Kupffer cells produce large amounts of mature IL-18. Similar to IL-1β, IL-18 is synthesized and stored as an inactive precursor form (pro-IL-18), and activation requires processing by caspase 1 in response to the appropriate signaling. It then exits the cell through a nontraditional secretory pathway. The IL-18 receptor (IL-18R) is composed of two subunits, IL-18Rα and IL-18Rβ, and is a member of the IL-1R superfamily, which is structurally similar in its cytoplasmic domains to the TLR.
One unique biologic property of IL-18 is the potential, in conjunction with IL-12, to promote the Th1 response to bacterial infection. At the same time, exogenous IL-18 can also enhance the Th2 response and Ig-mediated humoral immunity, as well as augment neutrophil function. Recent studies suggest that IL-18 therapy may hold promise as effective therapy in promoting immune recovery after severe surgical stress.80
Interferons were first recognized as soluble mediators that inhibited viral replication through the activation of specific antiviral genes in infected cells. Interferons are categorized into three types based on receptor specificity and sequence homology. The two major types, type I and type II, are discussed here.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


