the depth and degree of inflammation and the presence or absence of both intestinal and pseudopyloric metaplasia, separating them from atrophic changes8 (Table 13-2). This really formed the basis of all subsequent morphologic classifications of gastritis. In 1973, Strickland and Mackay classified gastritis based on detecting parietal cell (PC) antibodies, clarifying the etiology of autoimmune gastritis (AIG) (type A) despite the fact that these can develop in Helicobacter infected patients. It is associated with atrophic changes in body and fundic (oxyntic) mucosa. Antral predominant gastritis was type B. Glass and Pitchumon added type AB into Strickland-Mackay classification to encompass cases that did not fit type A or type B, essentially pangastritis.
Table 13-1 ABC Classification of Gastritis | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
antrum and corpus. It needs a minimum of two biopsies from the lesser and greater curvature of the respective gastric compartments as well as the incisura and any specific lesions identified (Fig. 13-2).13 On all occasions accurate grading depends on correctly oriented full-thickness mucosal biopsies. In practice, other than for academic studies, grading is rarely required.
Table 13-2 Gastritis Classification “Historical Prospective” | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
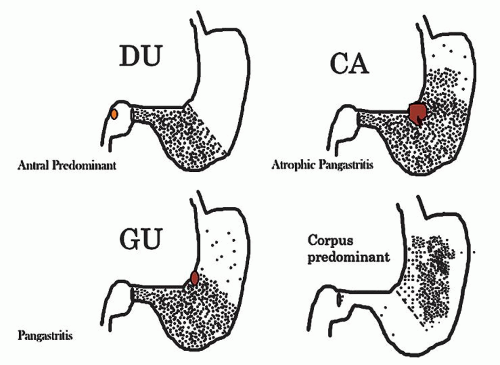 Figure 13-1. Prototypes of gastritis pattern predict disease outcome. In practice, all tend to have some degree of both antral and corpus inflammation. Top left: Duodenal ulcer (DU) patients have antral predominant inflammation with little corpus inflammation. Bottom left: Pangastritis is seen in gastric ulcer (GU) patients. Corpus mucosa is inflamed and often extends into the specialized mucosa but still tends to be antral predominant. Top right: Pangastritis with atrophy is seen in patients with the intestinal type of gastric adenocarcinoma (CA). Bottom right: Corpus-predominant gastritis is usually seen in AIG or end-stage Helicobacter infection. |
bowel disease (IBD), and side effects of medications (Table 13-4). As treatment depends on the cause, it is important to know the cause for appropriate management. Occasionally, it may be necessary to list possible etiologies for gastric inflammation, rather that reporting “nonspecific chronic inflammation”—which is an unnecessarily complex term as all inflammation is “nonspecific,” so these words can always be omitted from reports without deleterious effect. If it is specific, the cause (e.g., Helicobacter) should be stated.
Table 13-3 Gastritis Classification “Sydney System” | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
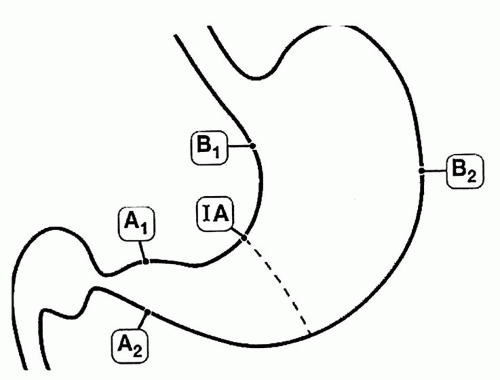 Figure 13-2. The updated Sydney biopsy protocol requires a minimum of two biopsies from the lesser and greater curvature of the respective gastric compartments as well as the incisura and any specific lesions identified. This identifies all of the patterns of gastritis illustrated in Figure 13-1, as well as estimating the extent of atrophy present, which often starts at the incisura/angulus (IA), affects the antrum (A1, A2), and then extends proximally to the oxyntic zone (B1, B2), so that, as antral inflammation extends proximally, biopsy site B1 is first affected, and B2 is the last site affected. |
vascular ectasia [GAVE], portal hypertension gastropathy, Dieulafoy, and hemorrhagic/shock) (Table 13-4). Graft versus host disease (GVHD) is usually normal endoscopically.
Table 13-4 Classification by Predominant Histologic Change | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
in practice it is often reported without qualifying it as acute or chronic. In some patients both are present together.
restituting, and appears attenuated as seen in any restitutional processes.
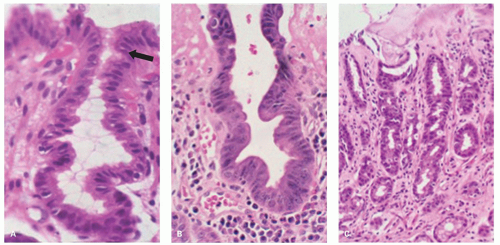 Figure 13-3. Reactive changes in gastric mucosa. A: Pit in which there is total mucin loss but nuclei are separated from each other. This is most marked superficially where the epithelial cells are more cuboidal and attenuated. Hints of mucin secretion are reappearing superficially (arrow) at the apex of the cell—an indication of maturation. Note the lack of any inflammation in the lamina propria in this biopsy. B: Similar features but there is more attenuation of epithelium superficially, and in the generative zone at the bottom nuclei are becoming stratified. The hyperchromatism associated with most dysplasias is absent. A modest chronic inflammatory infiltrate is present in the lamina propria but this disappears superficially. C: Chemical (NSAID) erosion. The attenuated epithelium is visible superficially with diffuse mucin depletion. Foveolar hyperplasia (corkscrewed pits) are visible, as is the normal architecture. At the surface the hyalinized zone is typical of NSAID damage. The lamina propria is largely empty indicating that this cannot be a Helicobacter-associated erosion. |
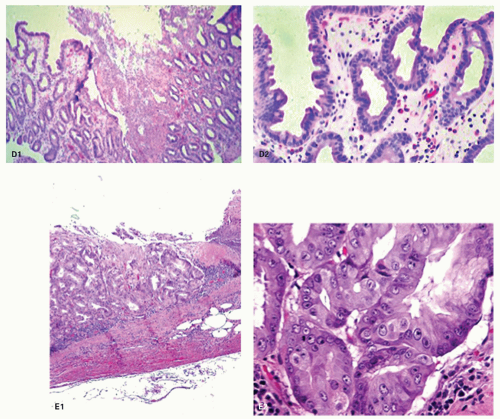 Figure 13-3. (Continued) D: D1. An erosion with almost a pseudomembranous appearance. D2. The adjacent mucosa has typical reactive changes and scattered eosinophils predominate. E1: Further NSAID erosion with the superficial hyalinized band that approaches the muscularis mucosae and E2: Very reactive nuclei, again most marked at the bases of the pits, nuclei remain separated but here have a prominent nucleolus. More superficially nuclei are even more widely separated indicating restitution. Note also that these nuclear changes do not correspond to intestinal, foveolar, or pyloric dysplasia. |
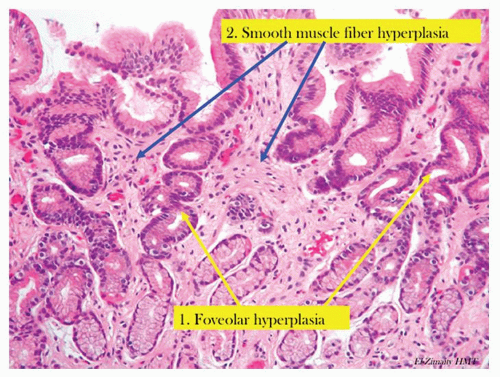 Figure 13-4. Chronic reactive gastropathy. The pits have foveolar hyperplasia, being elongated and have a corkscrew configuration (yellow arrows). There is hyperplasia of the smooth muscle fibers (blue arrow) that are normally found in the stomach. Note the lack of inflammation. |
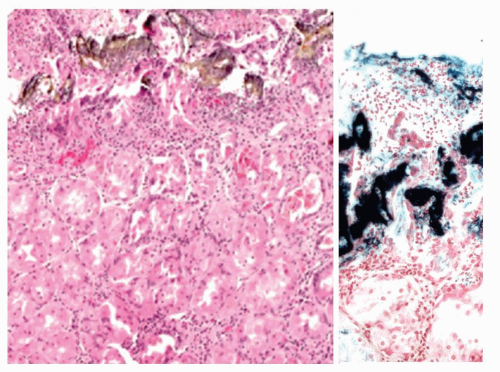 Figure 13-5. Reactive gastropathy. Iron medication may result in gastric mucosal necrosis (H&E stain) with iron encrustation (Perl’s stain—right) |
small mucin droplets at the surface. While “bottom-up” dysplasia (dysplasia maximal in the pit bases) does occur, it is quite rare, so the diagnosis of dysplasia should only be made if the diagnosis is absolutely clear, and ideally conforms to one of the usual forms of foveolar dysplasia (see following chapter). The adage that dysplasia should never be diagnosed in the presence of overlying or adjacent ulcers, erosions, or restituting epithelium unless absolutely clear is a good one. Making a diagnosis of dysplasia under these circumstances is fraught with danger. Unless there is absolutely no diagnostic uncertainty, it is usually best to rebiopsy the area following antisecretory therapy (e.g., PPIs) to ensure that the changes persist when the erosions have healed. Fortunately, even if dysplasia is diagnosed and graded, most can be visualized and treated endoscopically (Fig. 13-8).
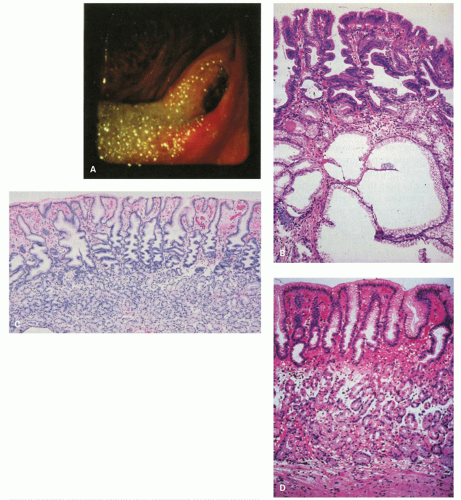 Figure 13-6. The postoperative stomach. A: Endoscopic view of a Billroth II stoma, which is typically red. Bile-stained fluid is refluxing into the gastric remnant. B: Biopsy specimen from the stoma of a Billroth II anastomosis. There is marked foveolar hyperplasia (corkscrew pattern), with minimal or no increase in the number of inflammatory cells. The epithelium in the surface and pits is dark and mucin depleted. Large intramucosal cysts are present. C: Fundic gland mucosa from the gastric body after Billroth II anastomosis. There is mild interfoveolar edema and marked foveolar hyperplasia with the corkscrew pattern, but an intact gland zone without increased numbers of inflammatory cells. D: Biopsy specimen from the greater curvature of the midbody region after Billroth II anastomosis. Many biopsy specimens in such patients simply show a thin fundic gland mucosa with a shallow epithelial gland zone, especially when the antrum has been removed as the gastrin drive for growth is lost. This specimen also shows subepithelial hemorrhage and edema in the interface between the pits and glands. It is not possible to exclude endoscope trauma as the cause of this finding. |
adjacent to alcohol- and NSAID-induced erosions27 in some patients without erosions on NSAIDs17 and in the mucosa at or near-healed gastric ulcer sites. Though, at times, it can be challenging, atypical reparative changes can be distinguished from dysplasia (intraepithelial neoplasia or dysplasia) as discussed in the previous section.
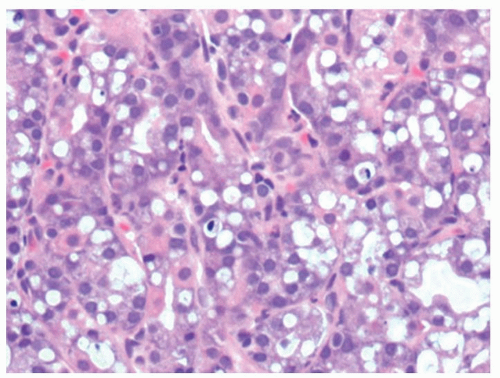 Figure 13-7. Vacuolated cells that are prominent in toxic states, in this patient the association was uremia. |
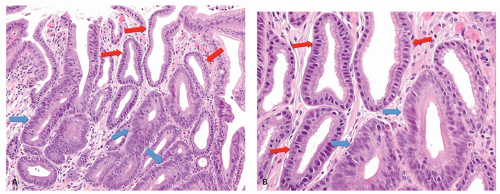 Figure 13-8. A: Reactive changes versus dysplasia. Typical reactive changes with mucin depletion, but widely spaced nuclei and superficially attenuated epithelium (red arrows). These contrast with the closely packed stratified nuclei in the dysplastic crypts (blue arrows). B: Detail of (A). Reactive changes (red arrows) versus low-grade dysplasia (blue arrows). |
tend to refer to events that happened at some point in the past, and it is unclear how long these changes take to reverse. If accompanied by acute changes of damage, then “reactive changes” covers both acute and chronic changes without the need to specify.
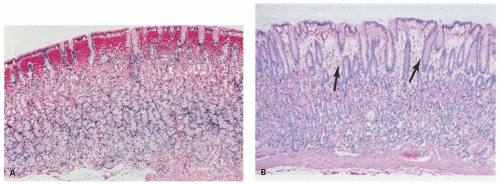 Figure 13-9. A: Biopsy specimen of a subepithelial hemorrhage in a patient with alcoholism. There is diffuse subepithelial hemorrhage across the full span of the fundic gland mucosa, but there is no inflammation present. B: Mucosal edema with an empty appearance of the interpit regions throughout the span of the biopsy (arrows). This is from a biopsy specimen adjacent to an area of subepithelial hemorrhage in a patient with alcoholism. |
was mild and was similar in severity in the lesions and the adjacent mucosa. In actively drinking alcoholic patients, the gastric mucosa may, in addition, exhibit the features of congestive gastropathy if there is portal hypertension. This is discussed in the next section.
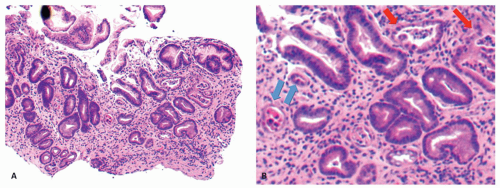 Figure 13-10. A: With chemotherapy, gastric injury is not uniform. There is architectural distortion and individual crypts are in various stages of repair. B: Individual crypts vary from some that are very attenuated and undergoing restitution (blue arrow) to others in which more typical regenerative changes can be found (red arrows). These are irregularly admixed with more normal-appearing pits. |
embolization to help control bleeding, accidental entry of selective intra-arterial radiotherapy (SIRT) beads, and vasculitis37, 38 or hypovolemic states are reported causes of erosive gastritis and gastric ulcers. There have also been isolated reports of patients with chronic gastric ulcers and erosions that healed after intestinal revascularization.39 The reported histology in these cases may lack the classic features of ischemia. In severe disease, epithelial and glandular cells are shed in the lumens of pits. Although this sounds innocuous, the mucous-producing cells can take on the appearances of signet ring cells, mimicking signet ring carcinoma (Fig. 13-11), analogous to similar lesions seen in pseudomembranous colitis. Another potential mimic of signet ring cells are the normal mucous-producing cells that appear in the oxyntic mucosa as it approaches the antrum. The polarity of these cells may appear abnormal, but these are terminally differentiated cells with no proliferative activity (Fig. 13-11E,F), while parietal cells can be shed into the lumen in oxyntic mucosa and raise the question of parietal cell carcinoma because of apparent disorderly sheets of cells (Fig. 13-12). The gastroduodenal subepithelial hemorrhages and erosions reported in some children with Henoch-Schonlein purpura might be due to vasculitis-induced mucosal ischemia.40
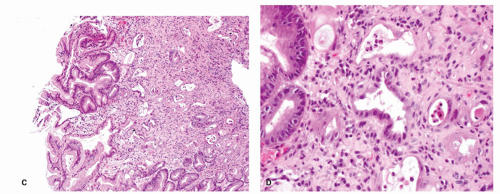 Figure 13-10. (Continued) C: Overview of second biopsy with more severe changes. The admixture of pits of different stages of degeneration and repair, some benign columnar and others lined by restituting epithelium. D: Severe chemotherapy changes with most glands being lined by restituting epithelium although focally they are more columnar. |
capillaries with fibrin thrombi and marked fibromuscular hyperplasia of the lamina propria41 (Fig. 13-13). Though it has been primarily described in the gastric antrum, proximal involvement has been reported.42
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


