KEY POINTS
Any patient admitted to a hospital because of peptic ulcer disease should be considered for lifelong acid suppression.
Lifelong acid suppressive medication may be equivalent to surgical vagotomy in preventing recurrent peptic ulcer or ulcer complications.
Roux-en-Y gastrojejunostomy should be avoided unless more than half of the stomach has been removed. Otherwise marginal ulceration and/or gastric stasis (Roux syndrome) may become problematic.
Gastric resection for peptic ulcer should be avoided in the asthenic or high-risk patient, if possible.
Many patients with locally advanced gastric cancer (T2b, T3, T4) are cured by an oncologically sound operation that includes wide margins and adequate lymphadenectomy.
Most patients with primary gastric lymphoma can be treated without gastric resection.
Gastric carcinoids should usually be removed either endoscopically or surgically. The surgeon should treat gastric carcinoid without hypergastrinemia (type III) as if it were malignant.
INTRODUCTION
The stomach is a remarkable organ with important digestive, nutritional, and endocrine functions. The stomach stores and facilitates the digestion and absorption of ingested food, and it helps regulate appetite. Treatable diseases of the stomach are common, and it is accessible and relatively forgiving. Thus, the stomach is a favorite therapeutic target. To provide intelligent diagnosis and treatment, the physician and surgeon must understand gastric anatomy, physiology, and pathophysiology. This includes a sound understanding of the mechanical, secretory, and endocrine processes through which the stomach accomplishes its important functions; and a familiarity with the common benign and malignant gastric disorders of clinical significance. The surgeon must also understand the indications, complications, and outcomes of procedures utilized to treat diseases of the stomach. The purpose of this chapter is to enhance the reader’s current understanding and familiarity with these concepts and topics. Some important milestones in the history of gastric surgery1,2,3,4,5,6 are listed in Table 26-1.
| DATE | EVENT | DATE | EVENT |
|---|---|---|---|
350 b.c. – 201 a.d. 1363 1586 1600–1700 1688 1737 1833 1869 1875 1879 1880 1880 1881 1884 1885 | Existence of gastric ulceration was acknowledged by Diocles of Carystos (350 b.c.), Celsus, and Galen (131–201 a.d.). Guy de Chauliac describes closure of gastric wound. Marcellus Donatus of Mantua describes gastric ulcer at autopsy. Reports of surgeons cutting stomach to remove foreign bodies. Muralto describes duodenal ulcer at autopsy. Morgagni describes both gastric and duodenal ulcer at autopsy. William Beaumont reports data recorded during his care of Alexis St. Martin who developed a gastric fistula from a left upper quadrant musket wound. Maury reportedly performs feeding gastrostomy to palliate esophageal stricture following consultation with Samuel D. Gross. Sidney Jones in London publishes the first successful gastrostomy for feeding. Paen performed distal gastrectomy and gastroduodenostomy. The patient died 5 d later. Rydygier resected a distal gastric cancer, and the patient died 12 h later. Billroth resects distal gastric cancer and performs gastroduodenostomy (Billroth I). Patient Therese Heller recovers and survives 4 mo. Anton Wolfler performs loop gastrojejunostomy to palliate an obstructing distal gastric cancer. Rydygier reports an unsuccessful gastrojejunostomy for benign gastric outlet obstruction. Billroth performs a successful distal gastrectomy and gastrojejunostomy (Billroth II) for gastric cancer. | 1886 1888 1892 1902 1891–1913 1920–1950 1943 1952 1953 1955 1957 1980–2000 1990–current 1995–current 2000–current | Heineke performs pyloroplasty. Mikulicz performs similar operation. Jaboulay describes bypassing the intact pylorus with gastroduodenostomy. Finney from Baltimore describes pyloroplasty technique. Different techniques of gastrostomy are described by Witzel (1891), Stamm (1894), and Janeway (1913). Subtotal gastrectomy grows popular as an operation for peptic ulcer. Von Haberer and Finsterer proponents. Dragstedt and Owen describe transthoracic truncal vagotomy to treat peptic ulcer disease. By the early 1950s, it is well recognized that some patients developed gastric stasis after this procedure, and transabdominal truncal vagotomy and drainage (pyloroplasty or gastrojejunostomy) become a standard ulcer operation. Farmer and Smithwick describe good results with truncal vagotomy and hemigastrectomy for peptic ulcer. Edwards and Herrington (Nashville) describe truncal vagotomy and antrectomy for peptic ulcer. Zollinger and Ellison describe the eponymous syndrome. Griffith and Harkins (Seattle) describe parietal cell vagotomy (highly selective vagotomy) for the elective treatment of peptic ulcer disease. Japanese surgeons and other surgical groups from East Asia demonstrate that more aggressive lymphadenectomy may improve survival in patients with gastric cancer. Evolving role of laparoscopic techniques in the treatment of surgical gastric disease. Dramatic increase in bariatric operations. Development of natural orifice translumenal endoscopic surgery. |
ANATOMY
The stomach is readily recognizable as the asymmetrical, pear-shaped, most proximal abdominal organ of the digestive tract (Fig. 26-1).7 The part of the stomach attached to the esophagus is called the cardia. Just proximal to the cardia at the gastroesophageal (GE) junction is the anatomically indistinct but physiologically demonstrable lower esophageal sphincter. At the distal end, the readily apparent pyloric sphincter connects the stomach to the proximal duodenum. The stomach is relatively fixed at these points, but the large midportion is quite mobile with the shorter lesser curvature on the right and the longer greater curvature on the left.
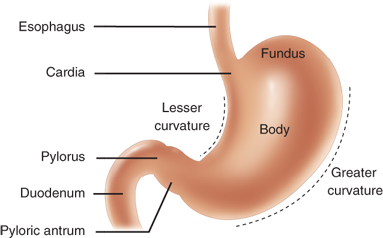
The superior-most part of the stomach is the distensible fundus, bounded superiorly by the diaphragm and laterally by the spleen. The angle of His is where the fundus meets the left side of the GE junction. Generally, the inferior extent of the fundus is considered to be the horizontal plane of the GE junction, where the body (corpus) of the stomach begins. The body of the stomach contains most of the parietal (oxyntic) cells, some of which are also present in the cardia and fundus. At the angularis, incisura the lesser curvature turns rather abruptly to the right, marking the anatomic beginning of the antrum, which comprises the distal 25% to 30% of the stomach.
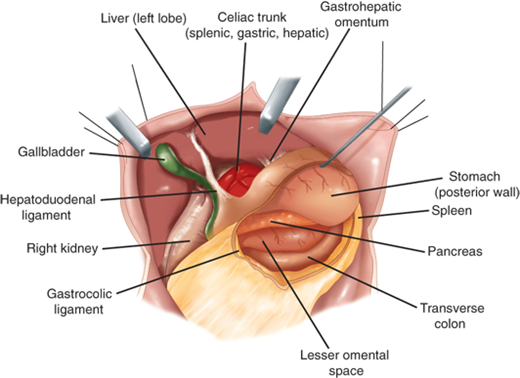
The organs that commonly abut the stomach are the liver, colon, spleen, pancreas, and occasionally the kidney (Fig. 26-2). The left lateral segment of the liver usually covers a large part of the anterior stomach. Inferiorly, the stomach is attached to the transverse colon by the gastrocolic omentum. The lesser curvature is tethered to the liver by the hepatogastric ligament, also referred to as the lesser omentum or pars flaccida. Posterior to the stomach is the lesser omental bursa and the pancreas.
Figure 26-2.
Anatomic relationships of the stomach. [Reproduced with permission from Mercer DW, Liu TH, Castaneda A: Anatomy and physiology of the stomach, in Zuidema GD, Yeo CJ (eds): Shackelford’s Surgery of the Alimentary Tract, 5th ed., Vol. II. Philadelphia: Saunders, 2002, p 3. Copyright Elsevier.]
The stomach is the most richly vascularized portion of the alimentary tube with ample blood flow and a dense intramural vascular anastomotic network. The large majority of the gastric blood supply is from the celiac axis via four named arteries (Fig. 26-3). The left and right gastric arteries form an anastomotic arcade along the lesser curvature, and the right and left gastroepiploic arteries form an arcade along the greater gastric curvature. The consistently largest artery to the stomach is the left gastric artery, which usually arises directly from the celiac trunk and divides into an ascending and descending branch along the lesser gastric curvature. Approximately 20% of the time, the left gastric artery supplies an aberrant vessel that travels in the gastrohepatic ligament (lesser omentum) to the left side of the liver. Rarely, this is the only arterial blood supply to this part of the liver, and inadvertent ligation may lead to clinically significant hepatic ischemia in this unusual circumstance. The more common smaller aberrant left hepatic artery may be ligated without significant consequences.
Figure 26-3.
Arterial blood supply to the stomach. a. = artery; v. = vein. [Reproduced with permission from Mercer DW, Liu TH, Castaneda A: Anatomy and physiology of the stomach, in Zuidema GD, Yeo CJ (eds): Shackelford’s Surgery of the Alimentary Tract, 5th ed., Vol. II. Philadelphia: Saunders, 2002, p 3. Copyright Elsevier.]
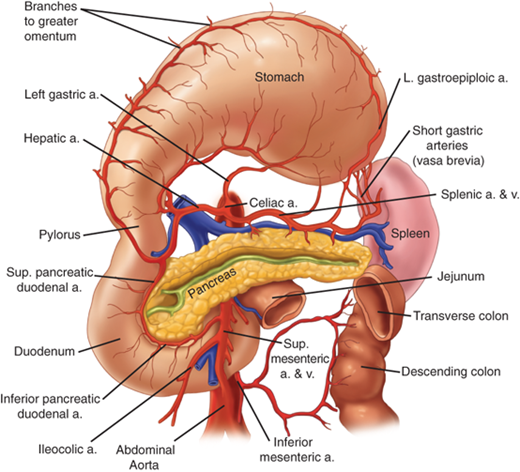
The second largest artery to the stomach is the right gastroepiploic artery, which arises consistently from the gastroduodenal artery behind the first portion of the duodenum. The left gastroepiploic artery arises from the splenic artery, and, together with the right gastroepiploic artery, forms the rich gastroepiploic arcade along the greater curvature. The right gastric artery usually arises from the hepatic artery near the pylorus and hepatoduodenal ligament, and runs proximally along the distal stomach. In the fundus along the proximal greater curvature, the short gastric arteries and veins arise from the splenic circulation. There also may be additional vascular branches to the proximal stomach from the phrenic and splenic circulation.
The veins draining the stomach generally parallel the arteries. The left gastric (coronary vein) and right gastric veins usually drain into the portal vein, though occasionally the coronary vein drains into the splenic vein. The right gastroepiploic vein drains into the superior mesenteric vein near the inferior border of the pancreatic neck, and the left gastroepiploic vein drains into the splenic vein.
The richness of the gastric blood supply and the extensiveness of the anastomotic connections have some important clinical implications, such as: a) At least two of the four named gastric arteries may be occluded or ligated with impunity. This is done routinely when the stomach is mobilized and pedicled on the right gastric and right gastroepiploic vessels to reach into the neck as an esophageal replacement (see Chapter 25) or during sleeve gastrectomy for weight loss when the gastroepiploic arcade is interrupted distally and resected (see Chapter 27), b) Following radical subtotal gastrectomy during which the right and left gastric arteries and both gastroepiploic arteries are all ligated, the gastric remnant is adequately supplied by short gastric arteries as long as the splenic artery is patent and intact; c) Angiographic control of gastric bleeding from deep ulcer or tumor often requires embolization of more than one feeding artery; d) Because of the rich venous interconnections in the stomach, a distal splenorenal shunt, which connects the distal end of the divided splenic vein to the side of the left renal vein, can effectively decompress esophagogastric varices in patients with portal hypertension.8,9
Generally speaking, the gastric lymphatics parallel the blood vessels (Fig. 26-4).10 The cardia and medial half of the corpus commonly drain to nodes along the left gastric and celiac axis. The lesser curvature side of the antrum usually drains to the right gastric and pyloric nodes, while the greater curvature half of the distal stomach drains to the nodes along the right gastroepiploic chain. The proximal greater curvature side of the stomach usually drains into nodes along the left gastroepiploic or splenic hilum. The nodes along both the greater and lesser curvature commonly drain into the celiac nodal basin. There is a rich anastomotic network of lymphatics that drain the stomach, often in a somewhat unpredictable fashion. Thus, a tumor arising in the distal stomach may give rise to positive lymph nodes in the splenic hilum. The rich intramural plexus of lymphatics and veins accounts for the fact that there can be microscopic evidence of malignant cells in the gastric wall at a resection margin that is several centimeters away from palpable malignant tumor. It also helps explain the not infrequent finding of positive lymph nodes which may be many centimeters away from the primary tumor, with closer nodes that remain negative.
Figure 26-4.
Lymph node stations draining the stomach according to the Japanese Research Society for Gastric Cancer. Stations 3 to 6 are commonly removed with D1 gastrectomy. Stations 1, 2, and 7 to 12 are commonly removed with D2 gastrectomy. [Reproduced with permission from Hermanek P, et al (eds): TNM Atlas: Illustrated Guide to the TNM/pTNM Classification of Malignant Tumours, 4th ed. Berlin: Springer-Verlag, 1997, p 82–83. Used with the permission of the International Union Against Cancer (UICC), Geneva, Switzerland.]
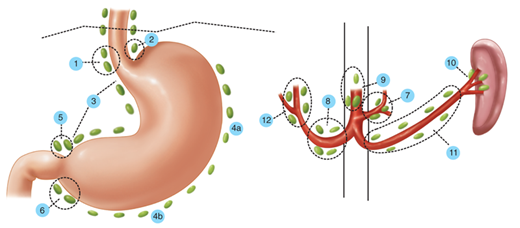
Extensive and meticulous lymphadenectomy is considered by many surgeons to be an important part of the operation for gastric cancer. Surgeons and pathologists have numbered the primary and secondary lymph node groups to which the stomach drains (see Fig. 26-4).11,12
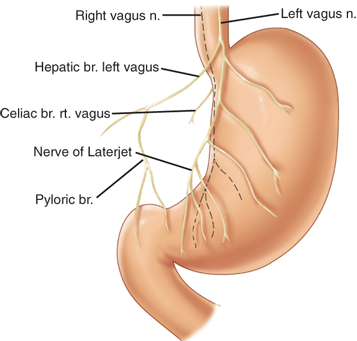
The vagus nerves provide the extrinsic parasympathetic innervation to the stomach, and acetylcholine is the most important neurotransmitter.13 From the vagal nucleus in the floor of the fourth cerebral ventricle, the vagus traverses the neck in the carotid sheath and enters the mediastinum, where it gives off the recurrent laryngeal nerve and divides into several branches around the esophagus. These branches come together again above the esophageal hiatus and form the left (anterior) and right (posterior) vagal trunks (mnemonic LARP). Near the GE junction the anterior vagus sends a branch (or branches) to the liver in the gastrohepatic ligament, and continues along the lesser curvature as the anterior nerve of Latarjet (Fig. 26-5). Similarly, the posterior vagus sends branches to the celiac plexus and continues along the posterior lesser curvature. The nerves of Latarjet send segmental branches to the body of the stomach before they terminate near the angularis incisura as the “crow’s foot,” sending branches to the antropyloric region. There may be additional branches to the distal stomach and pylorus that travel near the right gastric and/or gastroepiploic arteries. In 50% of patients, there are more than two vagal nerves at the esophageal hiatus. The branch that the posterior vagus sends to the posterior fundus is termed the criminal nerve of Grassi. This branch typically arises above the esophageal hiatus and is easily missed during truncal or highly selective vagotomy (HSV). Vagal fibers originating in the brain synapse with neurons in Auerbach’s myenteric plexus and Meissner’s submucosal plexus. In the stomach the vagus nerves affect secretion (including acid), motor function, and mucosal bloodflow and cytoprotection. They also play a role in appetite control and perhaps even mucosal immunity and inflammation. Most of the axons contained in the vagal trunks are afferent (i.e., carrying stimuli from the viscera to the brain).
The extrinsic sympathetic nerve supply to the stomach originates at spinal levels T5 through T10 and travels in the splanchnic nerves to the celiac ganglion. Postganglionic sympathetic nerves then travel from the celiac ganglion to the stomach along the blood vessels.
Neurons in the myenteric and submucosal plexuses constitute the intrinsic nervous system of the stomach. There may be more intrinsic gastric neurons than extrinsic neurons, but their function is poorly understood.
It is obviously an oversimplification (and incorrect) to think exclusively of the vagus as the cholinergic system and the sympathetic system as the adrenergic system of innervation. Although acetylcholine is an important neurotransmitter mediating vagal function, and epinephrine is important in the sympathetic nerves, both systems (as well as the intrinsic neurons) have various and diverse neurotransmitters, including cholinergic, adrenergic, and peptidergic (e.g., substance P and somatostatin).
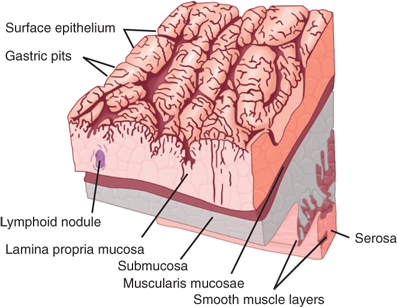
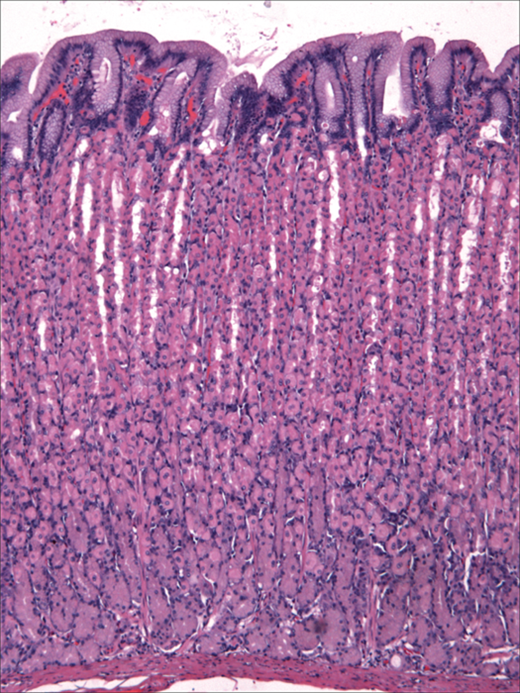
There are four distinct layers of the gastric wall: mucosa, submucosa, muscularis propria, and serosa (Fig. 26-6).7 The inner layer of the stomach is the mucosa, which is lined with columnar epithelial cells of various types. Beneath the basement membrane of the epithelial cells is the lamina propria, which contains connective tissue, blood vessels, nerve fibers, and inflammatory cells. Beneath the lamina propria is a thin muscle layer called the muscularis mucosa, the deep boundary of the mucosal layer of the gut. The epithelium, lamina propria, and muscularis mucosa constitute the mucosa (Fig. 26-7).14 The epithelium of the gastric mucosa is columnar glandular. A scanning electron micrograph shows a smooth mucosal carpet punctuated by the openings of the gastric glands. The gastric glands are lined with different types of epithelial cells, depending upon their location in the stomach (Fig. 26-8, Table 26-2).15,16 There are also endocrine cells present in the gastric glands. Progenitor cells at the base of the glands differentiate and replenish sloughed cells on a regular basis. Throughout the stomach, the carpet consists primarily of mucus-secreting surface epithelial cells (SECs) that extend down into the gland pits for variable distances. These cells also secrete bicarbonate and play an important role in protecting the stomach from injury due to acid, pepsin, and/or ingested irritants. In fact, all epithelial cells of the stomach (except the endocrine cells) contain carbonic anhydrase and are capable of producing bicarbonate.
| CELL TYPE | DISTINCTIVE ULTRASTRUCTURAL FEATURES | MAJOR FUNCTIONS |
|---|---|---|
| Surface-foveolar mucous cells | Apical stippled granules up to 1 μm in diameter | Production of neutral glycoprotein and bicarbonate to form a gel on the gastric luminal surface; neutralization of hydrochloric acida |
| Mucous neck cell | Heterogeneous granules 1–2 μm in diameter dispersed throughout the cytoplasm | Progenitor cell for all other gastric epithelial cells; glycoprotein production; production of pepsinogens I and II |
| Oxyntic (parietal) cell | Surface membrane invaginations (canaliculi); tubulovesicle structures; numerous mitochondria | Production of hydrochloric acid; production of intrinsic factor; production of bicarbonate |
| Chief cell | Moderately dense apical granules up to 2 μm in diameter; prominent supranuclear Golgi apparatus; extensive basolateral granular endoplasmic reticulum | Production of pepsinogens I and II, and of lipase |
| Cardiopyloric mucous cell | Mixture of granules like those in mucous neck and chief cells; extensive basolateral granular endoplasmic reticulum | Production of glycoprotein; production of pepsinogen II |
| Endocrine cells | See Figure 26-11 |
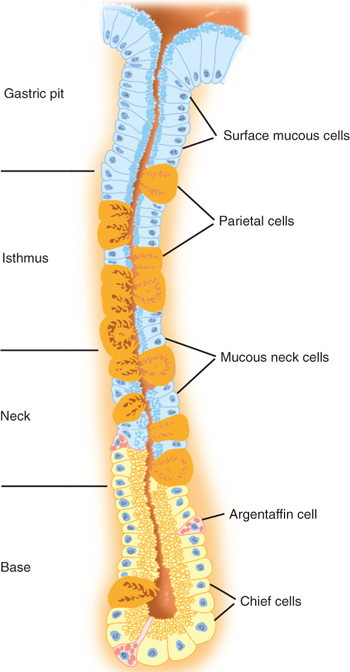
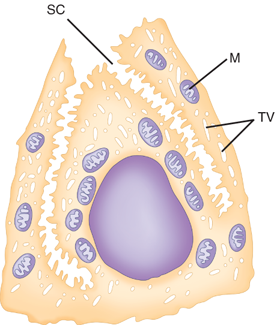
In the cardia, the gastric glands are branched and secrete primarily mucus and bicarbonate, but not much acid. In the fundus and body, the glands are more tubular and the pits are deep. Parietal and chief cells are common in these glands (Fig. 26-9). Histamine-secreting enterochromaffin-like (ECL) cells and somatostatin-secreting D cells are also found. Parietal cells secrete acid and intrinsic factor into the gastric lumen, and bicarbonate into the intercellular space. They have a characteristic ultrastructural appearance with secretory canaliculi (deep invaginations of the surface membrane), and cytoplasmic tubulovesicles containing the acid-producing apparatus H+/K+-ATPase (proton pump) (see Fig. 26-9). There are numerous mitochondria; in fact the parietal cell is the most mitochondria rich cell in the body. When the parietal cell is stimulated, the cytoplasmic tubulovesicles fuse with the membrane of the secretory canaliculus; when acid production ceases, the process is reversed. Arguably, parietal cells produce the only truly essential substance made by the stomach (i.e., intrinsic factor). Parietal cells tend to occupy the midportion of the gastric glands found in the corpus of the stomach.
Figure 26-9.
Ultrastructural features of the parietal (oxyntic) cell. SC = secretory canaliculus; M = mitochondria; TV = tubulovesicle. [Reproduced with permission from Antonioli DA, Madara JL: Functional anatomy of the gastrointestinal tract, in Ming S-C, Goldman H (eds): Pathology of the Gastrointestinal Tract, 2nd ed. Baltimore: Williams & Wilkins, 1998, p 13.]
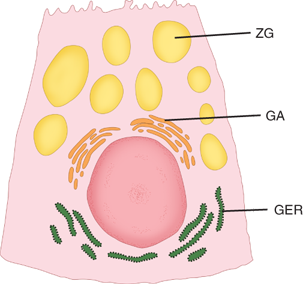
Chief cells (also called zymogenic cells) secrete pepsinogen I, which is maximally activated at a pH of 2.5. They tend to be clustered toward the base of the gastric glands and have a low columnar shape. Ultrastructurally, chief cells have the characteristics of protein-synthesizing cells: basal granular endoplasmic reticulum, supranuclear Golgi apparatus, and apical zymogen granules (Fig. 26-10). When stimulated, the chief cells produce two immunologically distinct proenzyme forms of pepsinogen: predominantly pepsinogen I and some pepsinogen II, most of which is produced by SECs. These proenzymes are activated in an acidic luminal environment.
Figure 26-10.
Ultrastructural features of the chief (zymogenic) cell. GA = Golgi apparatus; GER = granular endoplasmic reticulum; ZG = zymogen granule. [Reproduced with permission from Antonioli DA, Madara JL: Functional anatomy of the gastrointestinal tract, in Ming S-C, Goldman H (eds): Pathology of the Gastrointestinal Tract, 2nd ed. Baltimore: Williams & Wilkins, 1998, p 13.]
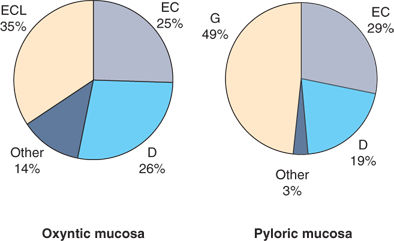
In the antrum, the gastric glands are again more branched and shallow, parietal cells are rare, and gastrin-secreting G cells and somatostatin-secreting D cells are present. A variety of hormone-secreting cells are present in various proportions throughout the gastric mucosa (Fig. 26-11).17 Histologic analysis suggests that in the normal stomach, 13% of the epithelial cells are oxyntic (parietal) cells, 44% are chief (zymogenic) cells, 40% are mucous cells, and 3% are endocrine cells. In general, the antrum produces gastrin but not acid, and the proximal stomach produces acid but not gastrin. The border between the corpus and antrum migrates proximally with age (especially on the lesser curvature side of the stomach).
Figure 26-11.
Endocrine cells of the stomach—proportion by site. D = d cell (somatostatin); EC = enterochromaffin cell; ECL = enterochromaffin-like cell (histamine); G = g cell (gastrin). [Reproduced with permission from Feldman M: Gastric secretion, in Feldman M, et al (eds):Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 7th ed. Philadelphia: Saunders, 2002, p 715. Copyright Elsevier.]
Deep to the muscularis mucosa is the submucosa, which is rich in branching blood vessels, lymphatics, collagen, various inflammatory cells, and nerve fibers and ganglion cells of Meissner’s autonomic submucosal plexus. The collagen-rich submucosa gives strength to GI anastomoses. The mucosa and submucosa are folded into the grossly visible gastric rugae, which tend to flatten out as the stomach becomes distended.
Below the submucosa is the thick muscularis propria (also referred to as the muscularis externa), which consists of an incomplete inner oblique layer, a complete middle circular layer (continuous with the esophageal circular muscle and the circular muscle of the pylorus), and a complete outer longitudinal layer (continuous with the longitudinal layer of the esophagus and duodenum). Within the muscularis propria is the rich network of autonomic ganglia and nerves that make up Auerbach’s myenteric plexus. Specialized pacemaker cells, the interstitial cells of Cajal (ICC), also are present.
The outer layer of the stomach is the serosa, also known as the visceral peritoneum. This layer provides significant tensile strength to gastric anastomoses. When tumors originating in the mucosa penetrate and breach the serosa, microscopic or gross peritoneal metastases are common, presumably from shedding of tumor cells that would not have occurred if the serosa had not been penetrated. In this way, the serosa may be thought of as an outer envelope of the stomach.
PHYSIOLOGY
The stomach stores food and facilitates digestion through a variety of secretory and motor functions. Important secretory functions include the production of acid, pepsin, intrinsic factor, mucus, and a variety of GI hormones. Important motor functions include food storage (receptive relaxation and accommodation), grinding and mixing, controlled emptying of ingested food, and periodic interprandial “housekeeping.”
Hydrochloric acid in the stomach hastens both the physical and (with pepsin) the biochemical breakdown of ingested food. In an acidic environment, pepsin and acid facilitate proteolysis. Gastric acid also inhibits the proliferation of ingested pathogens, which protects against both infectious gastroenteritis and intestinal bacterial overgrowth. Long-term acid suppression with proton pump inhibitors (PPIs) has been associated with an increased risk of community acquired Clostridium difficile colitis and other gastroenteritis, presumably because of the absence of this protective germicidal barrier.18,19
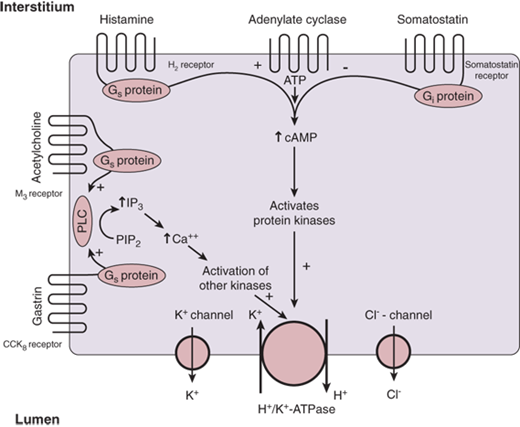
The parietal cell is stimulated to secrete acid (Fig. 26-12) when one or more of three membrane receptor types is stimulated by acetylcholine (from vagal nerve fibers), gastrin (from D cells), or histamine (from ECL cells).7,20,21 The enzyme H+/K+-ATPase is the proton pump. It is stored within the intracellular tubulovesicles and is the final common pathway for gastric acid secretion. When the parietal cell is stimulated, there is a cytoskeletal rearrangement and fusion of the tubulovesicles with the apical membrane of the secretory canaliculus. The heterodimer assembly of the enzyme subunits into the microvilli of the secretory canaliculus results in acid secretion, with extracellular potassium being exchanged for cytosolic hydrogen. Although electro-neutral, this is an energy requiring process because the hydrogen is secreted against a gradient of at least 1 million-fold, which explains why the parietal cell is packed with energy producing mitochondria. During acid production, potassium and chloride are also secreted into the apical secretory canaliculus through separate channels, providing potassium to exchange for H+ via the H+/K+-ATPase, and chloride to accompany the secreted hydrogen. At the basolateral membrane, the combined activity of various cotransporters and ion exchangers accomplishes intracellular pH regulation and electrolyte homeostasis.20
Figure 26-12.
Control of acid secretion in the parietal cell. ATP = adenosine triphosphate; cAMP = cyclic adenosine monophosphate; CCK = cholecystokinin; H2 = histamine 2; IP3 = inositol trisphosphate; PIP2 = phosphatidylinositol 4,5-bisphosphate; PLC = phospholipase C. [Reproduced with permission from Mercer DW, Liu TH, Castaneda A: Anatomy and physiology of the stomach, in Zuidema GD, Yeo CJ (eds): Shackelford’s Surgery of the Alimentary Tract, 5th ed., Vol. II. Philadelphia: Saunders, 2002, p 3. Copyright Elsevier.]
The normal human stomach contains approximately 1 billion parietal cells, and total gastric acid production is proportional to parietal cell mass. The potent acid-suppressing PPI drugs irreversibly interfere with the function of the H+/K+-ATPase molecule. These agents must be incorporated into the activated enzyme to be effective, and thus work best when taken before or during a meal (when the parietal cell is stimulated). When PPI therapy is stopped, acid secretory capability gradually returns (within days) as new H+/K+-ATPase is synthesized.
Gastrin, acetylcholine, and histamine stimulate the parietal cell to secrete hydrochloric acid (see Fig. 26-12). Gastrin binds to type B cholecystokinin (CCK) receptors, and acetylcholine binds to M3 muscarinic receptors. Both stimulate phospholipase C via a G-protein–linked mechanism leading to increased production of inositol trisphosphate from membrane bound phospholipids. Inositol trisphosphate stimulates the release of calcium from intracellular stores, which leads to activation of protein kinases and activation of H+/K+-ATPase. Histamine binds to the histamine 2 (H2) receptor, which stimulates adenylatecyclase, also via a G-protein–linked mechanism. Activation of adenylatecyclase results in an increase in intracellular cyclic adenosine monophosphate which activates protein kinases, leading to increased levels of phosphoproteins and activation of the proton pump. Somatostatin from mucosal D cells binds to membrane receptors and inhibits the activation of adenylatecyclase through an inhibitory G protein.
Food ingestion is the physiologic stimulus for acid secretion (Fig. 26-13). The acid secretory response that occurs after a meal is traditionally described in three phases: cephalic, gastric, and intestinal.22,23 The cephalic or vagal phase begins with the thought, sight, smell, and/or taste of food. These stimuli activate several cortical and hypothalamic sites (e.g., tractus solitarius, dorsal motor nucleus, and dorsal vagal complex), and signals are transmitted to the stomach by the vagal nerves. Acetylcholine is released, leading to stimulation of ECL cells and parietal cells. Although the acid secreted per unit of time in the cephalic phase is greater than in the other two phases, the cephalic phase is shorter. Thus, the cephalic phase accounts for no more than 30% of total acid secretion in response to a meal. Sham feeding (chewing and spitting) stimulates gastric acid secretion only via the cephalic phase, and results in acid secretion that is about half of that seen in response to IV pentagastrin or histamine.
Figure 26-13.
Physiologic control of acid secretion. ECL = enterochromaffin-like. [Reproduced with permission from Mercer DW, Liu TH, Castaneda A: Anatomy and physiology of the stomach, in Zuidema GD, Yeo CJ (eds): Shackelford’s Surgery of the Alimentary Tract, 5th ed., Vol. II. Philadelphia: Saunders, 2002, p 3. Copyright Elsevier.]
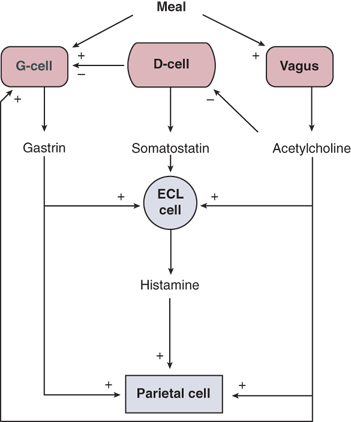
When food reaches the stomach, the gastric phase of acid secretion begins. This phase lasts until the stomach is empty, and accounts for about 60% of the total acid secretion in response to a meal. The gastric phase of acid secretion has several components. Amino acids and small peptides directly stimulate antral G cells to secrete gastrin, which is carried in the bloodstream to the parietal cells and stimulates acid secretion in an endocrine fashion. In addition, proximal gastric distention stimulates acid secretion via a vagovagal reflex arc, which is abolished by truncal or HSV. Antral distention also stimulates antral gastrin secretion. Acetylcholine stimulates gastrin release and gastrin stimulates histamine release from ECL cells.
The intestinal phase of gastric secretion is poorly understood. It is thought to be mediated by a hormone released from the proximal small bowel mucosa in response to luminal chyme. This phase starts when gastric emptying of ingested food begins, and continues as long as nutrients remain in the proximal small intestine. It accounts for about 10% of meal-induced acid secretion.
Interprandial basal acid secretion is 2 to 5 mEq hydrochloric acid per hour, about 10% of maximal acid output (MAO), and it is greater at night. Basal acid secretion probably contributes to the relatively low bacterial counts found in the stomach. Basal acid secretion is reduced 75% to 90% by vagotomy or H2 receptor blockade.
The pivotal role that ECL cells play in the regulation of gastric acid secretion is emphasized in Fig. 26-13. A large part of the acid stimulatory effects of both acetylcholine and gastrin are mediated by histamine released from mucosal ECL cells. H2 receptor knockout mice do not secrete acid in response to gastrin.20 This explains why the histamine 2 receptor antagonists (H2RAs) are such effective inhibitors of acid secretion, even though histamine is only one of three parietal cell stimulants. The mucosal D cell, which releases somatostatin, is also an important regulator of acid secretion. Somatostatin inhibits histamine release from ECL cells and gastrin release from antral G cells. The function of D cells is inhibited by Helicobacter pylori infection, and this leads to an exaggerated acid secretory response (see section Helicobacter pylori Infection).
The most potent physiologic stimulus for pepsinogen secretion from chief cells is food ingestion; acetylcholine is the most important mediator. Somatostatin inhibits pepsinogen secretion. Pepsinogen I is produced by chief cells in acid producing glands, whereas pepsinogen II is produced by SECs in both acid producing and gastrin producing (i.e., antral) glands. Pepsinogen is cleaved to the active pepsin enzyme in an acidic environment and is maximally active at pH 2.5, and inactive at pH >5, although pepsinogen II may be activated over a wider pH range than pepsinogen I. Pepsin catalyzes the hydrolysis of proteins and is denatured at alkaline pH.
Activated parietal cells secrete intrinsic factor in addition to hydrochloric acid. Presumably the stimulants are similar, but acid secretion and intrinsic factor secretion may not be linked. Intrinsic factor binds to luminal vitamin B12, and the complex is absorbed in the terminal ileum via mucosal receptors. Vitamin B12 deficiency can be life threatening, and patients with total gastrectomy or pernicious anemia (i.e., patients with no parietal cells) require B12 supplementation by a nonenteric route. Some patients develop vitamin B12 deficiency following gastric bypass, presumably because there is insufficient intrinsic factor present in the small proximal gastric pouch and oral B12 intake may be decreased. Under normal conditions, a significant excess of intrinsic factor is secreted, and acid-suppressive medication does not appear to inhibit intrinsic factor production and release.
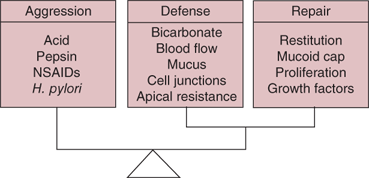
The stomach’s durable resistance to autodigestion by caustic hydrochloric acid and active pepsin is intriguing. Some of the important elements of gastric barrier function and cytoprotection are listed in Table 26-3.24,25 When these defenses break down, ulceration occurs. A variety of factors are important in maintaining an intact gastric mucosal layer. The mucus and bicarbonate secreted by SECs form an unstirred mucous gel with a favorable pH gradient. Cell membranes and tight junctions prevent hydrogen ions from gaining access to the interstitial space. Hydrogen ions that do break through are buffered by the alkaline tide created by basolateral bicarbonate secretion from stimulated parietal cells. Any sloughed or denuded SECs are rapidly replaced by migration of adjacent cells, a process known as restitution. Mucosal blood flow plays a crucial role in maintaining a healthy mucosa, providing nutrients and oxygen for the cellular functions involved in cytoprotection. “Back-diffused” hydrogen is buffered and rapidly removed by the rich blood supply. When “barrier breakers” such as bile or aspirin lead to increased back-diffusion of hydrogen ions from the lumen into the lamina propria and submucosa, there is a protective increase in mucosal blood flow. If this protective response is blocked, gross ulceration can occur. Important mediators of these protective mechanisms include prostaglandins, nitric oxide, intrinsic nerves, and peptides (e.g., calcitonin gene-related peptide, gastrin-releasing peptide [GRP], gastrin, and heat shock proteins). Sucralfate acts locally to enhance mucosal defenses. Protective reflexes involve afferent sensory neurons, and can be blocked by the application of topical anesthetics to the gastric mucosa, or the experimental destruction of the afferent sensory nerves. In addition to these local defenses, there are important protective factors in saliva, duodenal secretions, and pancreatic or biliary secretions.
Components Mucous barrier Bicarbonate secretion Epithelial barrier Hydrophobic phospholipids Tight junctions Restitution Microcirculation (reactive hyperemia) Afferent sensory neurons |
Mediators Prostaglandins Nitric oxide Epidermal growth factor Calcitonin gene-related peptide Hepatocyte growth factor Histamine Gastrin-releasing peptide |
Gastrin is produced by antral G cells and is the major hormonal stimulant of acid secretion during the gastric phase.13,26 A variety of molecular forms exist: big gastrin (34 amino acids; G34), little gastrin (17 amino acids; G17), and mini-gastrin (14 amino acids; G14). The large majority of gastrin released by the human antrum is G17. The biologically active pentapeptide sequence at the C-terminal end of gastrin is identical to that of CCK. Luminal peptides and amino acids are the most potent stimulants of gastrin release, and luminal acid is the most potent inhibitor of gastrin secretion. The latter effect is predominantly mediated in a paracrine fashion by somatostatin released from antral D cells. Gastrin-stimulated acid secretion is significantly blocked by H2 antagonists, suggesting that the principal mediator of gastrin-stimulated acid production is histamine from mucosal ECL cells (see Fig. 26-13). In fact, chronic hypergastrinemia is associated with hyperplasia of gastric ECL cells and, rarely, gastric carcinoid. Gastrin is trophic to gastric parietal cells and to other GI mucosal cells. Important causes of hypergastrinemia include pernicious anemia, acid-suppressive medication, gastrinoma, retained antrum following distal gastrectomy and Billroth II surgery, and vagotomy.
Somatostatin is produced by D cells located throughout the gastric mucosa. The predominant form in humans is somatostatin 14, though somatostatin 28 is present as well. The major stimulus for somatostatin release is antral acidification; acetylcholine from vagal nerve fibers inhibits its release. Somatostatin inhibits acid secretion from parietal cells and gastrin release from G cells. It also decreases histamine release from ECL cells. The proximity of the D cells to these target cells suggests that the primary effect of somatostatin is mediated in a paracrine fashion, but an endocrine (i.e., bloodstream) effect also is possible.
GRP is the mammalian equivalent of bombesin, a hormone discovered more than two decades ago in an extract of skin from a frog. In the antrum, GRP stimulates both gastrin and somatostatin release by binding to receptors on the G and D cells. There are nerve terminals ending near the mucosa in the gastric body and antrum, which are rich in GRP immunoreactivity. When GRP is given peripherally, it stimulates acid secretion, but when it is given centrally into the cerebral ventricles of animals, it inhibits acid secretion, apparently via a pathway involving the sympathetic nervous system. GRP is a mediator of gastroprotective increased mucosal blood flow in response to luminal irritants.
Leptin is a protein primarily synthesized in adipocytes. It is also made by chief cells in the stomach, the main source of leptin in the GI tract.26 Leptin works at least in part via vagally mediated pathways to decrease food intake in animals. Not surprisingly, leptin, a satiety signal hormone, and ghrelin, a hunger signal hormone, are both primarily synthesized in the stomach, an organ increasingly recognized as central to the mechanisms of appetite control.26,27
Ghrelin is a small peptide described in 1999 that is produced primarily in the stomach.28 Ghrelin is a potent secretagogue of pituitary growth hormone (but not adrenocorticotropic hormone, follicle-stimulating hormone, luteinizing hormone, prolactin, or thyroid-stimulating hormone). Ghrelin appears to be an orexigenic regulator of appetite (i.e., when ghrelin is elevated, appetite is stimulated, and when it is suppressed, appetite is suppressed). Resection of the primary source of this hormone (i.e., the stomach) may partly account for the anorexia and weight loss seen in some patients following gastric resection including sleeve gastrectomy (Fig. 26-14).29 The gastric bypass operation, a very effective treatment for morbid obesity, has been shown by some investigators to be associated with suppression of plasma ghrelin levels (and appetite) in humans (Fig. 26-15A).30,31
Figure 26-14.
Ghrelin levels are decreased after gastrectomy. (Reproduced with permission from Ariyasu H, Takaya K, Tagami T, et al: Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab. 86:4753, 2001.)
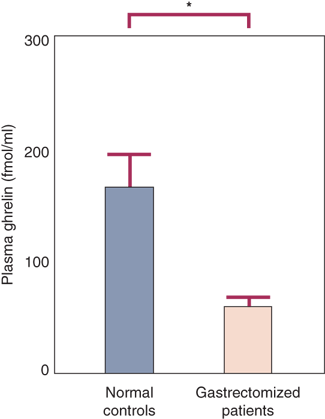
Figure 26-15.
A and B. Ghrelin secretion after bariatric surgery. Some investigators have suggested that ghrelin secretion is dramatically decreased after gastric bypass. Other groups have shown statistically insignificant changes in ghrelin levels after gastric bypass, (RYGBP) but significant decreases after sleeve gastrectomy (SG). A: green = gastric bypass; blue = obese controls; red = normal weight controls; B: blue = fasting; pink = postprandial. B. Top = RYGBP; B bottom = SG. (Fig. 26-15A reproduced with permission from Cummings DE, et al: Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 346:1623, 2002. Copyright ©2002 Massachusetts Medical Society. All rights reserved. Fig. 26-15B reproduced with permission from Karamanakos SN, et al: Weight loss, appetite suppression, and changes in fasting and postprandial ghrelin and peptide-YY levels after Roux-en-Y gastric bypass and sleeve gastrectomy: A prospective, double blind study. Ann Surg 247:401, 2008.)

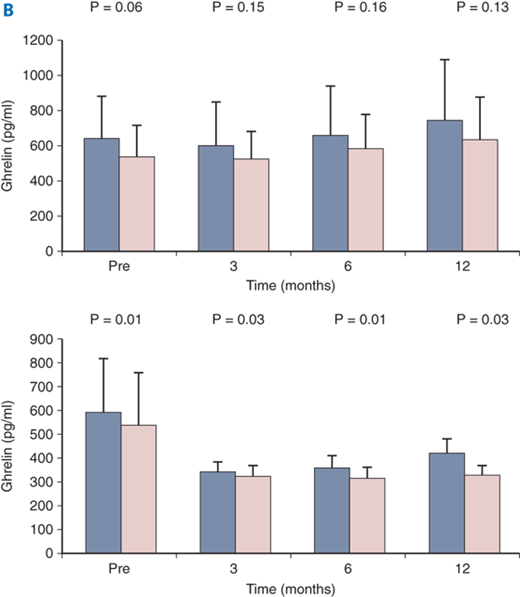
Other groups have failed to show a significant decrease in ghrelin levels following gastric bypass but have found such decreases following sleeve gastrectomy, another effective weight loss operation (Fig. 26-15B).31 Possibly subtle differences in operative technique, patient selection, and/or experimental conditions account for the findings that multiple studies have reported disparate results on the effect of bariatric surgery on ghrelin levels in obese patients. Obviously appetite control is complex with redundant and overlapping orexigenic and anorexigenic pathways and signals.26,27
Gastric motor function has several purposes.13,32,33,34 Interprandial motor activity clears the stomach of undigested debris, sloughed cells, and mucus. When feeding begins, the stomach relaxes to accommodate the meal. Regulated motor activity then breaks down the food into small particles and controls the output into the duodenum. The stomach accomplishes these functions by coordinated smooth muscle relaxation and contraction of the various gastric segments (proximal, distal, and pyloric). Smooth muscle myoelectric potentials are translated into muscular activity, which is modulated by extrinsic and intrinsic innervation and hormones. The mechanisms by which gastric distention is translated into a neurohormonal satiety signal have only been partially elucidated.26,27
The extrinsic parasympathetic and sympathetic gastric innervation was discussed previously under Innervation. The intrinsic innervation consists of ganglia and nerves that constitute the enteric nervous system (Fig. 26-16).35 There are a variety of neurotransmitters, which are generally grouped as excitatory (augment muscular activity) and inhibitory (decrease muscular activity). Important excitatory neurotransmitters include acetylcholine, the tachykinins, substance P, and neurokinin A. Important inhibitory neurotransmitters include nitric oxide (NO) and vasoactive intestinal peptide (VIP). Serotonin has been shown to modulate both contraction and relaxation. A variety of other molecules affect motility, including GRP, histamine, neuropeptide Y, norepinephrine, and endogenous opioids.
Figure 26-16.
Enteric nervous system. [Reproduced with permission from Chial HJ, Camilleri M: Motility disorders of the stomach and small intestine, in Friedman SL, McQuaid KR, Grendell JH (eds): Current Diagnosis and Treatment in Gastroenterology, 2nd ed. New York: McGraw-Hill, 2003, p 355. Copyright © The McGraw-Hill Companies, Inc.]
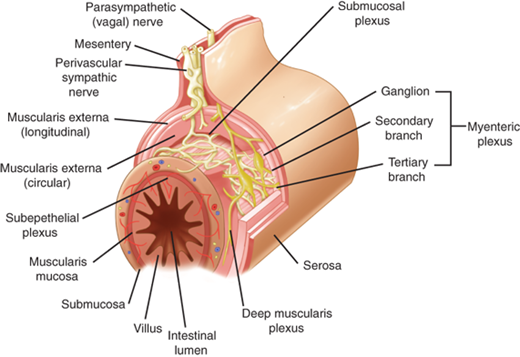
Specialized cells in the muscularis propria also are important modulators of GI motility. These cells, called interstitial cells of Cajal, are distinguishable histologically from neurons and myocytes, and appear to amplify both cholinergic excitatory and nitrergic inhibitory input to the smooth muscle of the stomach and intestine.36 They are thought to be the cell of origin for gastrointestinal stromal tumors (GISTs) which are the most common mesenchymal neoplasm in the GI tract.
13,36,37 In general, the proximal stomach serves a short-term food storage function and helps regulate basal intragastric tone, and the distal stomach mixes and grinds the food. The pylorus helps the latter process when closed, facilitating retropulsion of the solid food bolus back into the body of the stomach for additional breakdown. The pylorus opens intermittently to allow metered emptying of liquids and small solid particles into the duodenum.
Most of the motor activity of the proximal stomach consists of slow tonic contractions and relaxations, lasting up to 5 minutes. This activity is the main determinant of basal intragastric pressure, an important determinant of liquid emptying. Rapid phasic contractions may be superimposed on the slower tonic motor activity. When food is ingested, intragastric pressure falls as the proximal stomach relaxes. This proximal relaxation is mediated by two important vagovagal reflexes: receptive relaxation and gastric accommodation. Receptive relaxation refers to the reduction in proximal gastric tone associated with the act of swallowing. This occurs before the food reaches the stomach, and can be reproduced by mechanical stimulation of the pharynx or esophagus. Gastric accommodation refers to the proximal gastric relaxation associated with distention of the stomach. Accommodation is mediated through stretch receptors in the gastric wall and does not require esophageal or pharyngeal stimulation. Because both of these reflexes are mediated by afferent and efferent vagal fibers, they are significantly altered by truncal and highly selective vagotomy. Both these operations result in decreased gastric compliance, shifting the volume/pressure curve to the left. That is, interference with normal receptive relaxation and/or accommodation results in decreased gastric compliance, such that for any given amount of food or liquid ingested, the intragastric pressure is greater. This may increase the rate of liquid emptying, perhaps contributing to dumping symptoms after vagotomy.
NO and VIP are the principal mediators of proximal gastric relaxation. But a variety of other agents increase proximal gastric relaxation and compliance, including dopamine, gastrin, CCK, secretin, GRP, and glucagon. Proximal gastric tone also is decreased by duodenal distention, colonic distention, and ileal perfusion with glucose (ileal brake).
The distal stomach breaks up solid food and is the main determinant of gastric emptying of solids. Slow waves of myoelectric depolarization sweep down the distal stomach at a rate of about three per minute. These waves originate from the proximal gastric pacemaker, high on the greater curvature. The pacing cells may be interstitial cells of Cajal, which have been shown to have a similar function in the small intestine and colon. Most of these myoelectric waves are below the threshold for smooth muscle contraction in the quiescent state, and thus are associated with negligible changes in pressure. Neural and/or hormonal input, which increases the plateau phase of the action potential, can trigger muscle contraction, resulting in a peristaltic wave associated with the electrical slow wave, and of the same frequency (three per minute) (Fig. 26-17). It is possible that implantable gastric pacemakers benefit some patients with gastroparesis by favorably impacting this myoelectric coupling.
Figure 26-17.
The relationship between intracellular electrical activity and muscle cell contraction. Note that contractile activity is always associated with electrical activity, but the converse is not so. During mechanical quiescence, there are regular depolarizations that do not reach threshold. In the stimulated state, the threshold for contraction is reached, and motor activity is demonstrable. [Reproduced with permission from Kim CH: Electrical activity of the stomach: clinical implications. Mayo Clin Proc. 61:205, 1986.]
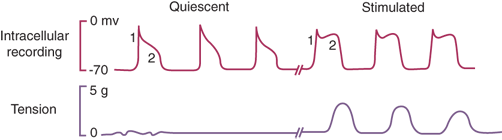
During fasting, distal gastric motor activity is controlled by the migrating motor complex (MMC), the “gastrointestinal housekeeper” (Fig. 26-18). The purported function of the MMC is to sweep along any undigested food, debris, sloughed cells, and mucus after the fed phase of digestion is complete. The MMC lasts approximately 100 minutes (longer at night, shorter during daytime) and is divided into four phases. Phase I (about half the length of the entire cycle) is a period of relative motor inactivity. High-amplitude muscular contractions do not occur in phase I of the MMC. Phase II (about 25% of the entire MMC cycle) consists of some irregular, high-amplitude, generally nonpropulsive contractions. Phase III, a period of intense, regular, (about three per minute) propulsive contractions, only lasts about 5 to 10 minutes. Most phase III complexes of the GI MMC begin in the stomach, and the frequency approximates that of the myoelectric gastric slow wave. Phase IV is a transition period.
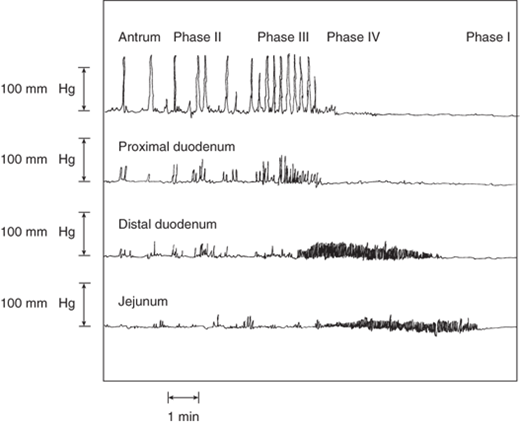
Figure 26-18.
Migrating motor complex, the fasting pattern of GI activity. During phase III of the migrating motor complex, effective peristaltic waves progress from the stomach to the distal small intestine. [Reproduced with permission from Rees WDW, et al: Human interdigestive and postprandial gastrointestinal motor and gastrointestinal hormone patterns. Dig Dis Sci. 27(4):321, 1982. Copyright 1982, with kind permission of Springer Science + Business Media.]
Neurohormonal control of the MMC is poorly understood, but it appears that different phases are regulated by different mechanisms. For example, vagotomy abolishes phase II of the gastric MMC but has little influence on phase III that persists even in the autotransplanted stomach, totally devoid of extrinsic neural input. This suggests that phase III is regulated by intrinsic nerves and/or hormones. Indeed, the initiation of phase III of the MMC in the distal stomach corresponds temporally to elevation in serum levels of motilin, a hormone produced in the duodenal mucosa. Resection of the duodenum abolishes distal gastric phase III in dogs, and resection of the duodenum in humans (e.g., with pancreaticoduodenectomy, the Whipple procedure) commonly results in early postoperative delayed gastric emptying. There are clearly motilin receptors on antral smooth muscle and nerves. Other modulators of gastric MMC activity include NO, endogenous opioids, intrinsic cholinergic and adrenergic nerves, and duodenal pH.
Feeding abolishes the MMC and leads to the fed motor pattern. The fed motor pattern of gastric activity starts within 10 minutes of food ingestion and persists until all the food has left the stomach. The neurohormonal initiator of this change is unknown, but CCK and the vagus appear to play some role: Sham feeding transiently induces antral motor activity resembling the fed motor pattern, and this is blocked by the CCK receptor antagonist loxiglumide. Gastric motility during the fed pattern resembles phase II of the MMC, with irregular but continuous phasic contractions of the distal stomach. During the fed state, about half of the myoelectric slow waves are associated with strong distal gastric contractions. Some are prograde and some are retrograde, serving to mix and grind the solid components of the meal. The magnitude of gastric contractions and the duration of the pattern are influenced by the consistency and composition of the meal.
The pylorus functions as an effective regulator of gastric emptying and an effective barrier to duodenogastric reflux. Bypass, transection, or resection of the pylorus may lead to uncontrolled gastric emptying of food and the dumping syndrome (see Postgastrectomy Problems). Pyloric dysfunction or disruption may also result in uncontrolled entry of duodenal contents into the stomach. Perfusion of the duodenum with lipids, glucose, amino acids, hypertonic saline, or hydrochloric acid results in closure of the pylorus and decreased transpyloric flow. Ileal perfusion with fat has the same effect. A variety of neurohumoral pathways are involved with these physiologic responses, and there is evidence that different pathways may be involved for different stimuli.
The pylorus is readily apparent grossly as a thick ring of muscle and connective tissue. The density of nerve tissue in the pyloric smooth muscle is several folds higher than in the antrum, with increased numbers of neurons staining positive for substance P, neuropeptide Y, VIP, and galanin. Interstitial cells of Cajal are is more closely associated with pyloric myocytes, and the myoelectric slow wave of the pylorus has the same frequency as that seen in the distal stomach. The motor activity of the pylorus is both tonic and phasic. During phase III of the MMC, the pylorus is open as gastric contents are swept into the duodenum. During the fed phase, the pylorus is closed most of the time. It relaxes intermittently, usually in synchronization with lower-amplitude, minor antral contractions. The higher-amplitude, more major antral contractions are usually met with a closed pylorus, facilitating retropulsion and further grinding of food.
Modulation of pyloric motor activity is complex. There is evidence of both inhibitory and excitatory vagal pathways. Some contractile vagal effects are mediated by opioid pathways, because they are blocked by naloxone. Electrical stimulation of the duodenum causes the pylorus to contract, whereas electrical stimulation of the antrum causes pyloric relaxation. Nitric oxide is an important mediator of pyloric relaxation. Other molecules that may play a physiologic role in controlling pyloric smooth muscle include serotonin, VIP, prostaglandin E1, and galanin (pyloric relaxation); and histamine, CCK, and secretin (pyloric contraction).
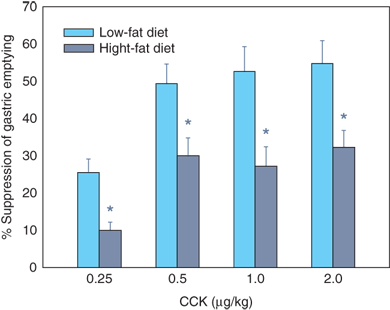
13 The control of gastric emptying is complex. In general, liquid emptying is faster than solid emptying. Osmolarity, acidity, caloric content, nutrient composition, and particle size are important modulators of gastric emptying. Stimulation of duodenal osmoreceptors, glucoreceptors, and pH receptors clearly inhibits gastric emptying by a variety of neurohumoral mechanisms. CCK has been consistently shown to inhibit gastric emptying at physiologic doses (Fig. 26-19). Recently, it has been noted that the anorexigenic hormone leptin, secreted by fat and gastric mucosa, inhibits gastric emptying, perhaps through the same pathway as CCK (which also has properties of a satiety hormone). The orexigenic hormone ghrelin has the opposite effect.
The gastric emptying of water or isotonic saline follows first-order kinetics, with a half emptying time around 12 minutes. Thus, if one drinks 200 mL of water, about 100 mL enters the duodenum by 12 minutes, whereas if one drinks 400 mL of water, about 200 mL enters the duodenum by 12 minutes. This emptying pattern of liquids is modified considerably as the caloric density, osmolarity, and nutrient composition of the liquid changes (Fig. 26-20). Up to an osmolarity of about 1 M, liquid emptying occurs at a rate of about 200 kcal per hour. Duodenal osmoreceptors and hormones (e.g., secretin and VIP) are important modulators of liquid gastric emptying. Generally, liquid emptying is delayed in the supine position.
Figure 26-20.
Nutrient composition and caloric density affect liquid gastric emptying. Glucose solution (purple circles), the least calorically dense, emptied the fastest. Other more calorically dense solutions, such as milk protein (green triangles) and peptide hydrolysates (red circles and blue triangles), emptied slower. [Reproduced with permission from Calbet JA, MacLean DA: Role of caloric content on gastric emptying in humans. J Physiol. 498:533, 1997.]

Traditionally, liquid emptying has been attributed to the activity of the proximal stomach, but it is probably more complicated than previously thought. Clearly, receptive relaxation and gastric accommodation play a role in gastric emptying of liquids. Patients with a denervated (e.g., vagotomized), resected, or plicated (e.g., fundoplication) proximal stomach have decreased gastric compliance and may show accelerated gastric emptying of liquids.
Some observations suggest an active role for the distal stomach in liquid emptying. For instance even if the proximal intragastric pressure is lower than duodenal pressure, normal gastric emptying of liquids can occur. Also, diabetic patients may have normal proximal gastric motor function and profoundly delayed gastric emptying of liquids. Indeed, antral contractile activity does correlate with liquid gastric emptying, and this distal gastric activity appears to vary with the nutrient composition and caloric content of the liquid meal. Depending on the circumstances, distal gastric motor activity can promote or inhibit gastric emptying of liquids. Distal gastrectomy and pyloric stenting both obviously interfere with distal gastric motor activity, and both accelerate the initial rapid phase of liquid gastric emptying.
Normally, the half-time of solid gastric emptying is less than 2 hours. Unlike liquids, which display an initial rapid phase followed by a slower linear phase of emptying, solids have an initial lag phase during which little emptying of solids occurs. It is during this phase that much of the grinding and mixing occurs. A linear emptying phase follows, during which the smaller particles are metered out to the duodenum. Solid gastric emptying is a function of meal particle size, caloric content, and composition (especially fat). When liquids and solids are ingested together, the liquids empty first. Solids are stored in the fundus and delivered to the distal stomach at constant rates for grinding. Liquids also are sequestered in the fundus, but they appear to be readily delivered to the distal stomach for early emptying. The larger the solid component of the meal, the slower the liquid emptying. Patients bothered by dumping syndrome are advised to limit the amount of liquid consumed with the solid meal, taking advantage of this effect. Three prokinetic agents are commonly used to treat delayed gastric emptying. Typical doses and mechanism of action are shown in Table 26-4.
DIAGNOSIS OF GASTRIC DISEASE
The most common symptoms of gastric disease are pain, weight loss, early satiety, and anorexia. Nausea, vomiting, bloating, and anemia also are frequent complaints. Several of these symptoms (pain, bloating, nausea, and early satiety) are often described by physicians as dyspepsia, synonymous with the common nonmedical term indigestion. Common causes of dyspepsia include gastroesophageal reflux disease (GERD) and disorders of the stomach, gallbladder, and pancreas. Although none of the above symptoms alone is specific for gastric disease, when elicited in the context of a careful history and physical examination, they point to a differential diagnosis, which can be refined with certain tests.
Patients with one or more of the alarm symptoms listed in Table 26-5 should undergo expeditious upper endoscopy. Esophagogastroduodenoscopy (EGD) is a safe and accurate outpatient procedure performed under conscious sedation.38 Smaller flexible scopes with excellent optics and a working channel are easily passed transnasally in the unsedated patient. Following an 8-hour fast, the flexible scope is advanced under direct vision into the esophagus, stomach, and duodenum. The fundus and GE junction are inspected by retroflexing the scope. To rule out cancer with a high degree of accuracy, all patients with gastric ulcer diagnosed on upper GI series or found at EGD should have multiple biopsy specimens of the base and rim of the lesion. Brush cytology also should be considered. Gastritis should be biopsied both for histologic examination and for a tissue urease test to rule out the presence of H. pylori. If Helicobacter infection is detected, it should probably be treated because of the etiologic association with peptic ulcers, mucosa-associated lymphoid tissue (MALT), and gastric cancer. The most serious complications of EGD are perforation (which is rare, but can occur anywhere from the cervical esophagus to the duodenum), aspiration, and respiratory depression from excessive sedation. Although EGD is a more sensitive test than double-contrast upper GI series, these modalities should be considered complementary rather than mutually exclusive.
Plain abdominal X-rays may be helpful in the diagnosis of gastric perforation (pneumoperitoneum) or delayed gastric emptying (large air-fluid level).
Double-contrast upper GI series may be better than EGD at elucidating gastric diverticula, fistula, tortuosity, stricture location, and size of hiatal hernia. Although there are radiologic characteristics of ulcers that suggest the presence or absence of malignancy, gastric ulcers always require adequate biopsy.
Usually, significant gastric disease can be diagnosed without these sophisticated imaging studies. However, one or the other should be part of the routine staging work-up for most patients with a malignant gastric tumor. Magnetic resonance imaging (MRI) may prove clinically useful as a quantitative test for gastric emptying, and may even hold some promise for the analysis of myoelectric derangements in patients with gastroparesis. Virtual gastroscopy using multi detector CT scan or MRI is not yet widely used but these techniques may prove useful for screening and staging of gastric disease.39,40,41 (Fig. 26-21).
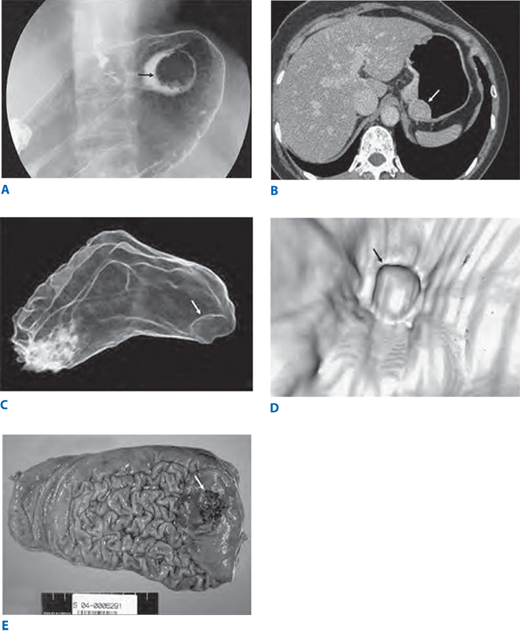
Figure 26-21.
Conventional double-contrast barium study (A) shows a focal protruding mass (arrow) on the gastric fundus. Axial computed tomographic scan (B) also shows a protruding polyp (arrow). The three dimensional computed tomographic gastrographic images in the transparent (C) and virtual endoscopic (D) modes show an elevated lesion on the gastric fundus (arrows). Photograph of the total gastrectomy specimen (E) shows a well-demarcated polypoid mass (arrow); this lesion was confirmed as early gastric carcinoma type I on microscopic examination (not shown). (Reproduced with permission from Shin KS, et al: Three-dimensional MDCT gastrography compared with axial CT for the detection of early gastric cancer. J Comput Assist Tomogr 31:741, 2007.)
Arteriography may be helpful in the occasional poor-risk patient with exsanguinating gastric hemorrhage, or in the patient with occult gastric bleeding.
Endoscopic ultrasound (EUS) is useful in the evaluation and management of some gastric lesions.42,43,44 Local staging of gastric adenocarcinoma with EUS is quite accurate, and this modality can be used to plan therapy. At some centers, patients with transmural and/or node positive adenocarcinoma of the stomach are considered for preoperative (neoadjuvant) chemoradiation therapy. EUS is the best way to clinically stage these patients locoregionally. Suspicious nodes can be sampled with EUS-guided endoscopic needle biopsy. Malignant tumors that are confined to the mucosa on EUS may be amenable to endoscopic mucosal resection (EMR). EUS also can be used to assess tumor response to chemotherapy. Submucosal masses are commonly discovered during routine EGD. Large submucosal masses should be resected because of the risk of malignancy, but observation may be appropriate for some small submucosal masses (e.g., lipoma or small GIST). There are endoscopic characteristics of benign and malignant mesenchymal tumors, and thus, EUS can provide reassurance, but no guarantee, that small lesions under observation are probably benign. Submucosal varices also can be assessed by EUS.
Analysis of gastric acid output requires gastric intubation, and it is performed infrequently nowadays. This test may be useful in the evaluation of patients with hypergastrinemia, including the Zollinger-Ellison syndrome (ZES), patients with refractory ulcer or GERD, and patients with recurrent ulcer after operation. Historically, gastric analysis was performed most commonly to test for the adequacy of vagotomy in postoperative patients with recurrent or persistent ulcer. Now this can be done by assessing peripheral pancreatic polypeptide levels in response to sham feeding.45 A 50% increase in pancreatic polypeptide within 30 minutes of sham feeding suggests vagal integrity.
Normal basal acid output (BAO) is greater than 5 mEq/h. MAO is the average of the two final stimulated 15-minute periods and is usually 10 to 15 mEq/h. Peak acid output is defined as the highest of the four stimulated periods. Patients with a gastrinoma commonly have a high BAO, often above 30 mEq/h, but consistently above 15 mEq/h unless there has been previous vagotomy or gastric resection. In patients with gastrinoma, the ratio of BAO to MAO exceeds 0.6. Normal acid output in the patient prescribed acid-suppressive medication usually means that the patient is noncompliant. To assess acid-secretory capacity in the absence of medication effect, H2 blockers and PPIs should be withheld before gastric analysis.
The standard scintigraphic evaluation of gastric emptying involves the ingestion of a test meal with one or two isotopes, and scanning the patient under a gamma camera. A curve for liquid and solid emptying is plotted, and the half-time calculated. Normal standards exist for each facility. Duodenogastric reflux can be quantitated by the IV administration of hepatobiliary iminodiacetic acid (HIDA scan), which is concentrated and excreted by the liver into the duodenum. Software allows a semiquantitative assessment of how much of the isotope refluxes into the stomach. Positron emission tomography (PET) scan or CT/PET scan may be useful in staging certain patients with gastric malignancy.
A variety of tests can help the clinician to determine whether the patient has active H. pylori infection.46 The predictive value (positive and negative) of any of these tests when used as a screening tool depends on the prevalence of H. pylori infection in the screened population. A positive test is quite accurate in predicting H. pylori infection, but a negative test is characteristically unreliable. Thus, in the appropriate clinical setting, treatment for H. pylori should be initiated on the basis of a positive test, but not necessarily withheld if the test is negative. Because of the association between H. pylori infection and gastric lymphoma and carcinoma, many clinicians recommend treating Helicobacter infection when the diagnosis is made.
A positive serologic test is presumptive evidence of active infection if the patient has never been treated for H. pylori. Histologic examination of an antral mucosal biopsy using special stains is the gold standard test. Other sensitive tests include commercially available rapid urease tests, which assay for the presence of urease in mucosal biopsy specimens (strong presumptive evidence of infection). Urease is an omnipresent enzyme in H. pylori strains that colonize the gastric mucosa. The labeled carbon-13 urea breath test has become the standard test to confirm eradication of H. pylori following appropriate treatment.47 In this test, the patient ingests urea labeled with nonradioactive 13C. The labeled urea is acted upon by the urease present in the H. pylori and converted into ammonia and carbon dioxide. The radiolabeled carbon dioxide is excreted from the lungs and can be detected in the expired air (Fig. 26-22). It also can be detected in a blood sample. The fecal antigen test also is quite sensitive and specific for active H. pylori infection and may prove more practical in confirming a cure.
Figure 26-22.
Labeled urea breath test to detect Helicobacter infection. (Reproduced with permission from Walsh JH, Peterson WL: The treatment of Helicobacter pylori infection in the management of peptic ulcer disease. N Engl J Med 333:984, 1995. Copyright ©1995 Massachusetts Medical Society. All rights reserved.)
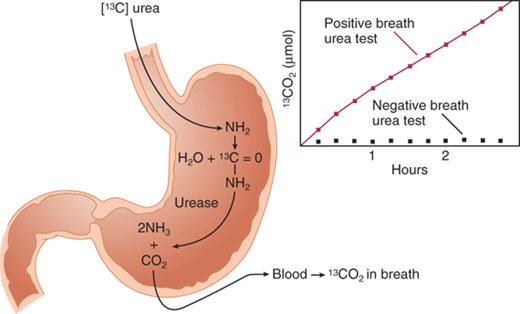
Antroduodenal motility testing and electrogastrography (EGG) are performed in specialized centers and may be useful in the evaluation of the patient with anomalous epigastric symptoms. EGG consists of the transcutaneous recording of gastric myoelectric activity. Antroduodenal motility testing is done with a tube placed transnasally or transorally into the distal duodenum. There are pressure-recording sensors extending from the stomach to the distal duodenum. The combination of these two tests together with scintigraphy provides a thorough assessment of gastric motility.
PEPTIC ULCER DISEASE
Peptic ulcers are focal defects in the gastric or duodenal mucosa that extend into the submucosa or deeper. They may be acute or chronic and, ultimately, are caused by an imbalance between mucosal defenses and acid/peptic injury (Fig. 26-23).48,49 Peptic ulcer remains a common outpatient diagnosis, but the number of physician visits, hospital admissions, and elective operations for PUD has decreased steadily and dramatically over the past three decades. Interestingly, the start of these trends all predated the use of H2 receptor blockers, or proton pump inhibitors, fiber-optic endoscopy, and highly selective vagotomy. However, the incidence of emergency surgery and the death rate associated with peptic ulcers has not decreased nearly so dramatically. These epidemiologic changes probably represent the net effect of several factors, including (beneficially) decreased prevalence of H. pylori infection, better medical therapy, and increased outpatient management; and (detrimentally) the use of NSAIDs and aspirin (with and without ulcer prophylaxis) in an aging population with multiple risk factors.
PUD is one of the most common GI disorders in the United States with a prevalence of about 2%, and a lifetime cumulative prevalence of about 10%, peaking around age 70 years.50 The costs of PUD, including lost work time and productivity, are estimated to be above $8 billion per year in the United States. In 1998, approximately 1.5% of all Medicare hospital costs were spent treating PUD, and the crude mortality rate for peptic ulcer was 1.7 per 100,000 individuals. Using the National Inpatient Sample, it can be estimated that the mortality rate in patients hospitalized in 2006 with duodenal ulcer was 3.7% compared to 2.1% for gastric ulcer.51 Recent studies have shown an increase in the rates of hospitalization and mortality in elderly patients for the peptic ulcer complications of bleeding and perforation. This may be due in part to the increasingly common use of NSAIDs and aspirin in this elderly cohort, many of whom also have H. pylori infection.
A variety of factors may contribute to the development of PUD. Although it is now recognized that the large majority of duodenal and gastric ulcers are caused by H. pylori infection and/or NSAID use17,52(Fig. 26-24), the final common pathway to ulcer formation is acid-peptic injury of the gastroduodenal mucosal barrier. Thus, the adage “no acid, no ulcer” remains true. Acid suppression heals both duodenal and gastric ulcers and prevents recurrence. In general H. pylori predisposes to ulceration, both by acid hypersecretion and by compromise of mucosal defense mechanisms. NSAID use causes ulcers predominantly by compromise of mucosal defenses. Duodenal ulcer was traditionally viewed as a disease of increased acid-peptic action on the duodenal mucosa, whereas gastric ulcer was viewed as a disease of weakened mucosal defenses. An increased understanding of peptic ulcer pathophysiology has blurred this overly simplistic distinction. Clearly, weakened mucosal defenses play a role in both duodenal and gastric ulcers and acid hypersecretion may result in a duodenal or gastric ulcer in the setting of normal mucosal defenses.
Figure 26-24.
“Causes” of peptic ulcer disease. Z.E. = Zollinger-Ellison syndrome. [Reproduced with permission from Spechler SJ: Peptic ulcer disease and its complications, in Feldman M (ed): Sleisinger and Fordtran’s Gastrointestinal and Liver Disease, 7th ed. Philadelphia: Saunders, 2002, p 747. Copyright Elsevier.]
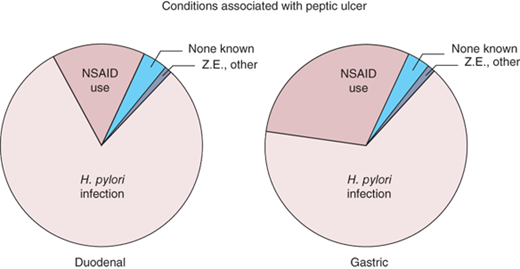
Elimination of H. pylori infection or NSAID use is important for optimal ulcer healing, and perhaps is even more important in preventing ulcer recurrence and/or complications. A variety of other diseases are known to cause peptic ulcer, including ZES (gastrinoma), antral G-cell hyperfunction and/or hyperplasia, systemic mastocytosis, trauma, burns, and major physiologic stress. Other causative agents include drugs (all NSAIDs, aspirin, and cocaine), smoking, and psychologic stress. In the United States, probably more than 90% of serious peptic ulcer complications can be attributed to H. pylori infection, NSAID use, and/or cigarette smoking.
With specialized flagella and a rich supply of urease, H. pylori is uniquely equipped for survival in the hostile environment of the stomach.53,54,55 About 50% of the world’s population is infected with H. pylori, a major cause of chronic gastritis. The same sequence of inflammation to metaplasia to dysplasia to carcinoma, that is well known to occur in the esophagus from reflux-induced inflammation (and in the colon from inflammatory bowel disease), is now increasingly well recognized to occur in the stomach with Helicobacter-induced gastritis. The influence of prolonged acid suppression with PPIs or H2RAs on these esophagogastric processes is largely unknown. Helicobacter also clearly has an etiologic role in the development of gastric lymphoma.
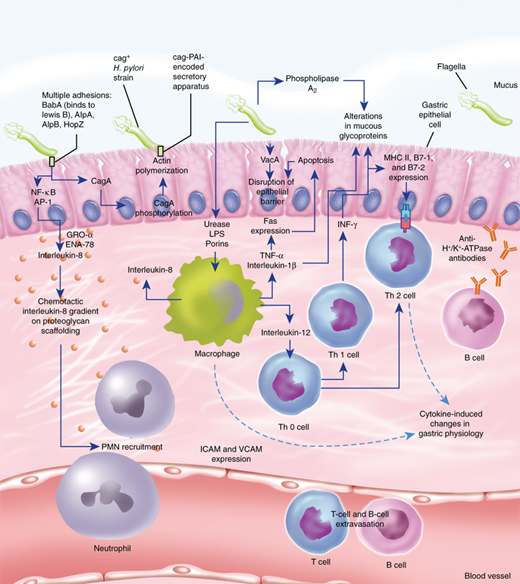
The organism possesses the enzyme urease, which converts urea into ammonia and bicarbonate, thus creating an environment around the bacteria that buffers the acid secreted by the stomach. The ammonia is damaging to the surface epithelial cells. Mutant strains of H. pylori that do not produce urease are unable to colonize the stomach. The organism lives in the mucus layer atop the gastric SECs, and some attach to these cells (Fig. 26-25).53,54 Apparently Helicobacter strains that lack flagella are unable to navigate through the unstirred mucus layer to get to the apical membrane of the SEC for attachment, and are nonpathogenic. One of the mechanisms by which Helicobacter causes gastric injury may be through a disturbance in gastric acid secretion. This is due, in part, to the inhibitory effect that H. pylori exerts on antral D cells that secrete somatostatin, a potent inhibitor of antral G-cell gastrin production. H. pylori infection is associated with decreased levels of somatostatin, decreased somatostatin messenger RNA production, and fewer somatostatin-producing D cells. These effects are probably mediated by H. pylori–induced local alkalinization of the antrum (antral acidification is the most potent antagonist to antral gastrin secretion), and H. pylori–mediated increases in other local mediators and cytokines. The result is hypergastrinemia and acid hypersecretion (Fig. 26-26).49 This hypergastrinemia presumably leads to the parietal cell hyperplasia seen in many patients with duodenal ulcer. The acid hypersecretion and the antral gastritis are thought to lead to antral epithelial metaplasia in the postpyloric duodenum. This duodenal metaplasia allows H. pylori to colonize the duodenal mucosa and, in these patients, the risk of developing a duodenal ulcer increases 50-fold. When H. pylori colonizes the duodenum, there is a significant decrease in acid-stimulated duodenal bicarbonate release. When H. pylori infection is successfully treated, acid secretory physiology tends to normalize. Other mechanisms whereby H. pylori can induce gastroduodenal mucosal injury include the production of toxins (vacA and cagA), local elaboration of cytokines (particularly interleukin-8) by infected mucosa, recruitment of inflammatory cells and release of inflammatory mediators, recruitment and activation of local immune factors, and increased apoptosis (Fig. 26-27).54,55 The net effect is a weakening of mucosal defenses.The mechanism by which the helicobacter organism avoids recognition and destruction by the mucosal immune system is a topic of interest and active research.56
Figure 26-25.
Helicobacter pylori closely adherent to the cell membrane (top), and spiral-shaped H. pylori attached to epithelial surface and surrounding microvilli (bottom). In the image on the bottom, the bacterial flagella can be seen arising from the upper pole of the bacterium. (From Parsonnet J: Clinician-discoverers—Marshall, Warren, and H. pylori. N Engl J Med 353:2421, 2005. Photomicrographs courtesy of Manuel Amieva, Stanford University.)

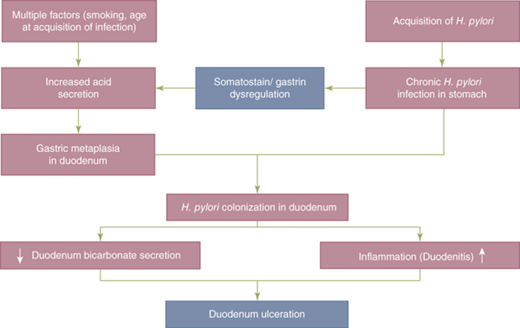
Figure 26-27.
Pathogen-host interactions in the pathogenesis of Helicobacter pylori infection. ICAM = intercellular adhesion molecule-1; IFN-γ = interferon-γ; LPS = lipopolysaccharide; NF-κB = nuclear factor κB; PAI = pathogenicity island; PMN = polymorphonuclear neutrophil; TNF-α = tumor necrosis factor alpha; VCAM = vascular cell adhesion molecule. (Reproduced with permission from Suerbaum S, Michetti P: Helicobacter pylori infection. N Engl J Med 347:1175, 2002. Copyright ©2002 Massachusetts Medical Society. All rights reserved.)
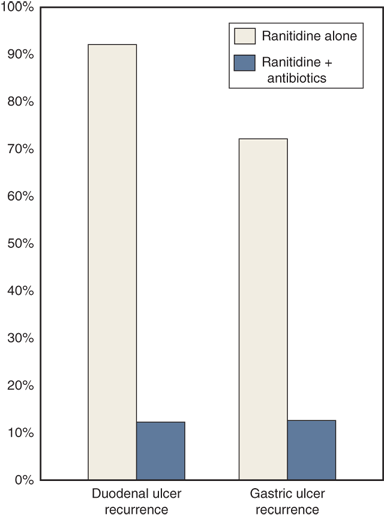
The evidence supporting the central role of H. pylori in the pathophysiology of PUD is strong. Patients with H. pylori infection and antral gastritis are three and one-half times more likely to develop PUD than patients without H. pylori infection. Up to 90% of patients with duodenal ulcers, and 70% to 90% of patients with gastric ulcers, have H. pylori infection. It is clear from multiple randomized prospective studies that curing H. pylori infection dramatically alters the natural history of PUD, decreasing the recurrent ulcer rate from more than 75% in patients treated with a course of acid-suppressive therapy alone (in whom H. pylori is not eradicated) to less than 20% in patients treated with a course of antibacterial therapy (Fig. 26-28).57
Figure 26-28.
Helicobacter treatment dramatically decreases the recurrence rate of duodenal and gastric ulcer. (Data from Graham DY, Lew GM, Klein PD, et al: Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized controlled study. Ann Intern Med 116:705, 1992.)
Obviously, other factors are involved in the etiology of PUD because many patients colonized with H. pylori do not get peptic ulcer disease, and many patients with peptic ulcer disease do not have H. pylori infection. These observations notwithstanding, it is clear from a variety of well-designed laboratory, clinical, and epidemiologic studies that H. pylori is indubitably an important factor in the development and recurrence of PUD. H. pylori also plays an etiologic role in gastric cancer and lymphoma.54
A variety of abnormalities related to mucosal acid exposure have been described in patients with duodenal ulcer (Fig. 26-29).58 Although duodenal ulcer patients as a group have a higher mean BAO and mean MAO compared to normal controls, many duodenal ulcer patients have basal and peak acid outputs in the normal range, and there is no correlation between acid secretion and the severity of the ulcer disease. As a group, duodenal ulcer patients produce more acid than normal controls in response to any known acid secretory stimulus. Although they usually have normal fasting serum gastrin levels, DU patients often produce more gastric acid at any given dose of gastrin than controls. Considering that many duodenal ulcer patients do produce excessive gastric acid, it has been argued that a “normal” fasting gastrin level in these patients is inappropriately high, and that there is an impaired feedback mechanism, especially in light of the apparently increased sensitivity of the parietal cell mass to gastrin. Many of these long-standing observations now seem reasonable in light of recently gained understanding of the perturbations in acid and gastrin secretion associated with H. pylori infection. Some patients with duodenal ulcer also have increased rates of gastric emptying that deliver an increased acid load per unit of time to the duodenum. Finally, the buffering capacity of the duodenum in many patients with duodenal ulcer is compromised due to decreased duodenal bicarbonate secretion.
Figure 26-29.
Frequency of physiologic abnormalities in patients with duodenal ulcer (DU). HCO3 = bicarbonate; MAO = maximal acid output. [Reproduced with permission from Del Valle J, Chey WD, Scheiman JM: Acid peptic disorders, in Yamada T, et al (eds): Textbook of Gastroenterology, 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2003, p 1321.]
In patients with gastric ulcer, acid secretion is variable. Currently, five types of gastric ulcer are described, although the original Johnson classification contained three types (Fig. 26-30).59 The most common, Johnson type I gastric ulcer, is typically located near the angularis incisura on the lesser curvature, close to the border between antral and corpus mucosa. Patients with type I gastric ulcer usually have normal or decreased acid secretion. Type II gastric ulcer is associated with active or quiescent duodenal ulcer disease, and type III gastric ulcer is prepyloric ulcer disease. Both type II and type III gastric ulcers are associated with normal or increased gastric acid secretion. Type IV gastric ulcers occur near the GE junction, and acid secretion is normal or below normal. Type V gastric ulcers are medication induced and may occur anywhere in the stomach. Patients with gastric ulcers may have weak mucosal defenses that permit an abnormal amount of injurious acid back-diffusion into the mucosa. Duodenogastric reflux may play a role in weakening the gastric mucosal defenses, and a variety of components in duodenal juice, including bile, lysolecithin, and pancreatic juice, have been shown to cause injury and inflammation in the gastric mucosa. NSAIDs and aspirin have similar effects. Although chronic gastric ulcer usually is associated with surrounding gastritis, it is unproven that the latter leads to the former.
Figure 26-30.
Modified Johnson classification for gastric ulcer. I. Lesser curve, incisura. II. Body of stomach, incisura + duodenal ulcer (active or healed). III. Prepyloric. IV. High on lesser curve, near gastroesophageal junction. V. Medication-induced (NSAID/acetylsalicylic acid), anywhere in stomach. [Reproduced with permission from Fisher WE, Brunicardi FC: Benign gastric ulcer, in Cameron JL (ed): Current Surgical Therapy, 9th ed. Philadelphia: Mosby Elsevier, 2008, p 81. Copyright Elsevier.]
Chronic use of NSAIDs (including aspirin) increases the risk of peptic ulcer disease about 5-fold and upper GI bleeding about 4-fold.60,61 Complications of PUD (specifically hemorrhage and perforation) are much more common in patients taking NSAIDs. More than half of patients who present with peptic ulcer hemorrhage or perforation report the recent use of NSAIDs, including aspirin. Many of these patients remain asymptomatic until they develop these life-threatening complications.
The overall risk of significant serious adverse GI events in patients taking NSAIDs is more than three times that of controls (Table 26-6). This risk increases to five times in patients more than age 60 years old. In elderly patients taking NSAIDs, the likelihood that they will require an operation related to a GI complication is 10 times that of the control group, and the risk that they will die from a GI cause is about four and one-half times higher. This problem is put into perspective when one realizes that approximately 20 million patients in the United States take NSAIDs on a regular basis; probably more regularly take aspirin. Persons who take NSAIDs also have a higher hospitalization rate for serious GI events than those who do not.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


