|
Typical Clinical Features |
Microscopic Features |
Ancillary Investigations |
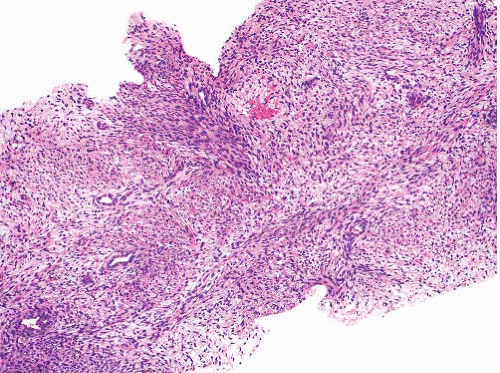
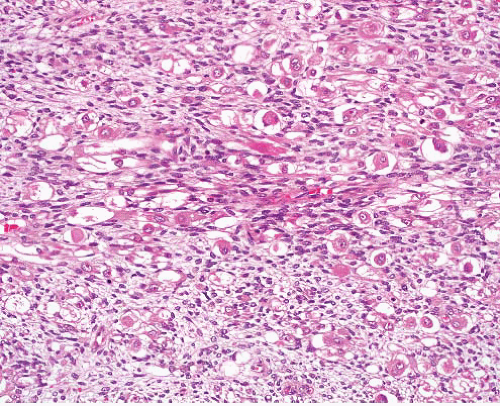
Malignant peripheral nerve sheath tumor |
M > F
In neurofibromatosis type 1 or sporadic, axial or in limbs
Origin in nerve or neurofibroma |
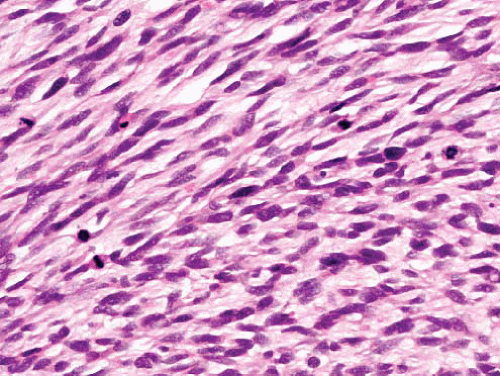
Variably cellular and myxoid, fascicular pattern
Nuclei wavy, buckled, or lanceolate with one blunt end
Palisading, neuroid whorls, vascular wall involvement, collagenous “rosettes” |
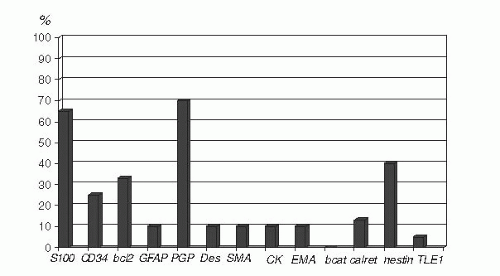
S100 protein+ (65%), occasional GFAP+, CK+ (but not usually CK7 or CK19), SOX10+ |
Cellular schwannoma |
F > M
Middle age
Paravertebral in retroperitoneum or pelvis, can erode bone
Also submucosal in nose, stomach, or intestine |
Thick capsule, subcapsular lymphoid aggregates
Fascicles of cells with eosinophilic cytoplasm, focal pleomorphism, occasional mitoses
Lacks Antoni A and B areas
Lymphocytes, clusters of foamy cells, thick-walled vessels, hemosiderin |
S100 protein diffusely+, CD34+ focally, GFAP+ focally, occasional CK+, CD117− |
Clear cell sarcoma |
Extremities especially lower limb, young adults
Subcutaneous or deep soft tissue, related to tendons or aponeuroses |
Nests of rounded or spindled cells, round nuclei with central nucleolus, clear or granular cytoplasm
Occasional multinucleated cells, melanin pigment in some |
S100 protein+, HMB45+ and melan-A+, other markers negative
t(12;22)(q13;q12) EWSR1-ATF1 fusion |
Perivascular epithelioid tumor |
Intra-abdominal, uterine, extremities (rarely) |
Nests of ovoid or spindled cells with clear or granular cytoplasm, delicate fibrous septa
Malignant variants often epithelioid |
SMA+, HMB45+, melan-A+, desmin±, CD117±, S100 protein+ rarely |
Synovial sarcoma |
Slowly growing mass, any location but mostly around knee |
Sheets of uniform short spindle cells with ovoid nuclei and minimal cytoplasm so that nuclei appear to overlap
Many mast cells
Poorly differentiated synovial sarcoma is a small round cell tumor |
CK+, EMA+, bcl-2+, CD99+, S100 protein+, TLE1+, CD34−, CD117−
t(X;18)(p11;q11)
SS18-SSX fusions |
Spindle epithelial tumor with thymus-like differentiation |
Adolescents or young adults, in or adjacent to thyroid
Can metastasize |
Biphasic pattern with mucous glands in spindle cell component
Can resemble synovial sarcoma |
CK5/6+, CK7+, CK20−, bcl-2+, CD99+, TLE1+ rarely
Genetic features of synovial sarcoma absent |
Spindle cell (sarcomatoid) carcinoma |
Related to epithelial surface or viscus
Previous carcinoma at same site |
Pleomorphic spindled tumor cells in sheets, sometimes areas of epithelial morphology or overlying dysplasia
Nested reticulin pattern
Rare osteochondroid or skeletal muscle differentiation |
CK+, EMA+, CD34−, SMA±, desmin−, h-caldesmon− |
Sarcomatoid mesothelioma |
Sheet-like mass involving peritoneal surface or omentum
Can present as metastasis
History of asbestos exposure |
Fascicles of pleomorphic spindle cells, tapered nuclei, scanty cytoplasm, mitoses, necrosis
Desmoplastic or hyalinized stroma
Epithelioid component in some |
CK+, calretinin+, EMA+ occasionally, D2-40+ occasionally, CD34−, bcl-2− |
Solitary fibrous tumor |
Circumscribed mass in subcutis, deep soft tissue, abdomen, retroperitoneum, thorax, viscera |
Circumscribed, not usually encapsulated
Distinct cellular and fibrous areas, focal myxoid stroma, patternless short spindle cells
Hemangiopericytomatous pattern focally
Malignant variant has hypercellularity, mitoses >4 per 10 hpf, necrosis |
STAT6+, CD34+, bcl-2+, CD99+ |
Adult fibrosarcoma |
Older adults, rare
Deep soft tissue, pelvis retroperitoneum |
Herringbone fascicles of spindle cells with tapered nuclei |
Some subsets CD34+, other markers negative |
Infantile fibrosarcoma |
Mostly first year, <4 y
Deep soft tissue, rapid growth |
Sheets or fascicles of ovoid cells, hemangiopericytomatous pattern, hemorrhage |
SMA+
Occasional CK+, ALK−
t(12;15)(p13;q25) ETV6–NTRK3 fusion |
Inflammatory myofibroblastic tumor |
Mesentery, retroperitoneum, lung, other sites |
Fascicular, myxoid, sclerosing patterns, mostly bland myofibroblasts, occasional atypical polygonal cells
Marked inflammation, especially plasma cells, often present in clumps |
SMA+, ALK+ 55%
ALK and other gene rearrangements |
Low-grade myofibrosarcoma |
Head and neck, extremities, retroperitoneum, bone, infiltrative mass, recurs |
Cellular fascicles infiltrate muscle
Myofibroblastic morphology, mostly uniform, but focal nuclear atypia is diagnostic
Occasional necrosis in higher grade tumors |
SMA+, desmin±, h-caldesmon− |
Leiomyosarcoma |
F > M
Limbs, head and neck, retroperitoneum, bowel wall or wall of vessel including inferior vena cava, renal vein |
Fascicles at right angles
Cells elongated with eosinophilic cytoplasm and nontapered nuclei
Paranuclear vacuoles
Myxoid change, fibrosis |
SMA+, desmin+, h-caldesmon+
Occasional CK+ (dot) |
Low-grade fibromyxoid sarcoma |
Skin, deep soft tissue, limbs/girdles |
Fibromatosis-like areas, swirling fibromyxoid transitions, cellular myxoid areas without pleomorphism
Some nuclei lozenge shaped, no nucleoli
Giant collagenous rosettes |
Occasionally SMA+, or CD34+, mostly lacks markers
t(7;16)(q34;p11)
FUS-CREB3L2 or FUSCREB3L1 fusion, rarely t(11;22(p13;q12),
EWSR1-CREB3L1 |
Fibromatosis, desmoid type |
Deep soft tissue, limbs, head and neck, body cavities |
Parallel myofibroblasts evenly dispersed in uniform collagen, slit-like and thickwalled vessels, mast cells
Normal mitoses acceptable but not nuclear atypia or necrosis |
SMA+, nuclear betacatenin+, CD34− |
Perineurioma |
Skin, subcutis, most locations
Subset in colon
Intraneural, sclerosing, reticular, and plexiform variants |
Spindle cells with long thin nuclei and very long slender terminal processes, in fascicles or perivascular whorls
Fibrous or myxoid stroma
Usually no atypia |
EMA+, claudin-1+, CD34±, beta-catenin−, S100 protein− |
Sclerosing epithelioid fibrosarcoma |
Deep soft tissue, limbs/girdles, head and neck
Can involve or arise in bone |
Multinodular, focal calcification
Cellular islands in dense fibrosis
Nests of ovoid cells, clear cytoplasm, or single files simulating carcinoma
Fibrosarcoma-like spindle cell areas in many cases |
Occasional and variable expression of bcl-2, EMA, CK, S100 protein. No specific immunophenotype
Some have molecular genetic features of low-grade fibromyxoid sarcoma |
Spindle cell rhabdomyosarcoma |
Children and adolescents in neck or paratestis
More aggressive subset in adults |
Fascicles of spindle cells resembling smooth muscle, scattered rhabdomyoblasts
In adults, high-grade spindle cell sarcoma in fascicles with variable myoid differentiation |
Desmin+, myogenin+ (nuclear), MyoD1+ (nuclear)
H-caldesmon− |
Follicular dendritic cell sarcoma |
Omentum, gastrointestinal tract, liver, spleen, soft tissue |
Sheets, whorls, and fascicles of ovoid cells
Prominent nuclear membranes, speckled chromatin
Intimate admixture of lymphocytes
Rarely giant cells, pleomorphism, necrosis |
CD21/35+, CD23+, S100 protein±, EMA+, D2-40+, fascin+, clusterin+, desmoplakin+, CD45− |
Angiosarcoma |
Older adults, head and neck, deep soft tissue |
Infiltrative, variably cellular nodules, focal vasoformation, epithelioid areas, hemorrhage |
CD31+, CD34+, FLI-1+ (nuclear), ERG+, HHV8−. |
Kaposi sarcoma |
Skin, soft tissue, lymph nodes, viscera |
Curved fascicles, mild pleomorphism
Intercellular hemorrhage, hyaline globules |
HHV8+ (nuclear), CD31+, CD34+, ERG+, D2-40+, CD117+ |
Intranodal myofibroblastoma |
Inguinal lymph node, rarely submandibular node
Very rarely recurs |
Rim of lymph node
Solid, hemorrhagic cut surface
Cellular fascicles of slender spindle cells with nuclear palisading
Hyaline-walled vessels, amianthoid fibers, hemorrhage, hemosiderin |
SMA+, other markers negative |
Intimal sarcoma |
Older adults
Pulmonary trunk or artery, thoracic or abdominal aorta |
Undifferentiated pleomorphic sarcoma
Spindle and polygonal cells
Myxoid change
Can show myoid, endothelial, or osseous differentiation |
SMA+ focally, other markers according to differentiation |