KEY POINTS
Sarcomas are a heterogeneous group of tumors that can occur throughout the body and encompass more than 50 subtypes with distinct histologic lines of differentiation.
Approximately two thirds of soft tissue sarcomas arise in the extremities; the remaining one third is distributed between the retroperitoneum, trunk, abdomen, head, and neck.
Multimodality treatment, including surgical resection, radiation therapy, and, in selected cases, systemic chemotherapy, has been applied to patients with locally advanced, high-grade, extremity sarcomas.
Overall 5-year survival rate for patients with all stages of soft tissue sarcoma is 50% to 60%.
These rare tumors account for less than 1% of cancer in adults (estimated 10,000 cases per year in the United States) and represent 15% of cancers in children.
The treatment algorithm for soft tissue sarcomas depends on tumor stage, site, and histology.
Of the patients who die of sarcoma, most will succumb to metastatic disease in the lungs, which 80% of the time occurs within 2 to 3 years of the initial diagnosis.
Progress in the understanding of soft tissue sarcoma biology is crucial for the development of new treatments.
INTRODUCTION
Sarcomas are a heterogeneous group of neoplasms that arise predominantly from cells of the embryonic mesoderm. While the majority of sarcomas are soft tissue sarcomas, other types of sarcoma include bone sarcomas (osteosarcoma, chondrosarcoma, and rare bone tumors like chordoma, angiosarcoma, and leiomyosarcoma of bone) and Ewing’s sarcoma/peripheral primitive neuroectodermal tumor, which can occur either in the bone or in the soft tissues. The primary focus of this chapter is soft tissue sarcomas. Most primary soft tissue sarcomas originate in an extremity (50%–60%); the next most common sites are the trunk (19%), retroperitoneum (15%), and head and neck (9%). The anatomic site of a primary sarcoma influences treatment and outcome.1
Soft tissue sarcomas include more than 50 histologic subtypes (Table 36-1). Historically, the most common subtypes in adults (excluding Kaposi’s sarcoma) were malignant fibrous histiocytoma (28%), liposarcoma (15%), leiomyosarcoma (12%), synovial sarcoma (10%), and malignant peripheral nerve sheath tumor (6%).2 Today, malignant fibrous histiocytoma is classified as either leiomyosarcoma, pleomorphic undifferentiated sarcoma, myxofibrosarcoma, or dedifferentiated liposarcoma based on cellular differentiation and genetics. Embryonal/alveolar rhabdomyosarcomas are the most common soft tissue sarcomas of childhood, whereas pleomorphic rhabdomyosarcoma occurs predominantly in adults, and although it shares part of the name, it has a different biology and should not be treated as a pediatric sarcoma.
| HISTOLOGIC SUBTYPES | NO. | % |
|---|---|---|
| Liposarcoma | 188 | 15 |
| Leiomyosarcoma | 148 | 12 |
| Unclassified sarcoma | 140 | 11 |
| Synovial sarcoma | 125 | 10 |
| Malignant peripheral nerve sheath tumor | 72 | 6 |
| Rhabdomyosarcoma | 60 | 5 |
| Fibrosarcoma | 38 | 3 |
| Ewing sarcoma | 25 | 2 |
| Angiosarcoma | 25 | 2 |
| Osteosarcoma | 14 | 1 |
| Epithelioid sarcoma | 14 | 1 |
| Chondrosarcoma | 13 | 1 |
| Clear cell sarcoma | 12 | 1 |
| Alveolar soft part sarcoma | 7 | 1 |
| Malignant hemangiopericytoma | 5 | 0.4 |
During the past 25 years, patients with extremity sarcomas have been treated with a multimodality approach, which has led to some improvements in survival, local control, and quality of life.3 However, patients with abdominal sarcomas continue to have high rates of recurrence and poor overall survival.4 The overall 5-year survival rate for patients with all stages of soft tissue sarcoma is 50% to 60%. Of the patients who die of sarcoma, most succumb to lung metastasis, which 80% of the time occurs within 2 to 3 years after initial diagnosis.
INCIDENCE
In the United States in 2012, approximately 11,280 new cases of soft tissue sarcoma were diagnosed, and 3900 deaths were attributable to this disease.5 The true incidence of sarcoma is thought to be higher than reported, and gastrointestinal stromal tumors (GISTs) likely account for an additional 5000 new sarcoma cases per year.1 Overall, sarcomas affect 5 to 6 individuals per 100,000 inhabitants per year,6 accounting for less than 1% of all malignancies in adults and 15% of malignancies in children.7
EPIDEMIOLOGY
Except for malignant peripheral nerve sheath tumors in patients with neurofibromatosis, sarcomas do not seem to result from progression or dedifferentiation of a benign soft tissue tumor. While most sarcomas are of unknown cause, a few sarcoma subtypes have been observed in settings suggesting etiology.
External radiation therapy is a rare but well-established risk factor for soft tissue sarcoma that may be associated with radiation-induced mutations of the p53 gene.8 The incidence of sarcoma among patients who are often treated with radiation for cancer of the breast, cervix, ovary, testes, or lymphatic system is 8 to 50 times the general-population risk.9,10 In a review of 160 patients with postirradiation sarcomas, the most common histologic types were osteogenic sarcoma, malignant fibrous histiocytoma, angiosarcoma, and lymphangiosarcoma.9 The risk of developing a sarcoma increased with radiation dose, and the median time between radiation therapy and diagnosis of sarcoma was 10 years.9 A recent review of 44 patients with radiation-induced sarcomas identified between 1989 and 2009 noted that the average period from initial radiation treatment to diagnosis was 16 years and that radiation-induced sarcomas occurred most commonly in patients treated for breast cancer (36% of the patients in the series) and lymphoma (34% of the patients in the series).11 The 5-year overall survival rate for patients presenting without metastasis was 44%.
Exposure to herbicides such as phenoxyacetic acids and to wood preservatives containing chlorophenols has been linked to an increased risk of soft tissue sarcoma.12 Several chemical carcinogens, including thorium oxide (Thorotrast), vinyl chloride, and arsenic, have been associated with hepatic angiosarcomas.13
Although patients with sarcoma often report a history of trauma, no causal relationship has been established. More often, a minor injury calls attention to a pre-existing tumor.
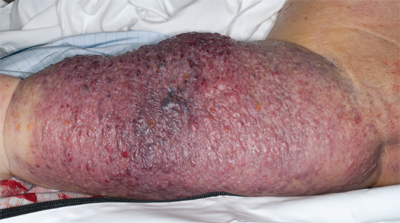
In 1948, Stewart and Treves first described the association between chronic lymphedema after axillary dissection and subsequent lymphangiosarcoma (Fig. 36-1).14 Lymphangiosarcoma has been estimated to occur in 0.07% of patients who undergo axillary node dissection.15 It also has been reported to occur after filarial infections and in the lower extremities of patients with congenital lymphedema.16,17 Lymphangiosarcoma is generally an aggressive tumor; average survival of patients with lymphangiosarcoma is 19 months.18
MOLECULAR PATHOGENESIS
Sarcomas can be broadly classified into three groups according to the genetic events underlying their development: specific translocations or gene amplification, defining oncogenic mutations, and complex genomic rearrangements.19 In general, sarcomas resulting from identifiable molecular events tend to occur in younger patients with histology suggesting a clear line of differentiation. The identifiable molecular events include point mutations, translocations causing overexpression of an autocrine grow factor, and oncogenic fusion transcription factor producing a cellular environment prone to malignant transformation. In contrast, sarcomas without identifiable genetic changes or expression profile signatures tend to occur in older patients and exhibit pleomorphic cytology and p53 dysfunction.20 Improved understanding of the molecular pathogenesis of sarcomas has revealed several potential targets against which investigators are working to develop subtype-specific targeted therapy.
To date, translocations have been identified in 14 subtypes of soft tissue sarcoma, accounting for 20% to 30% of all sarcomas21 (Table 36-2). Translocations result in in-frame gene fusion, which in turn results in fused products encoding oncoproteins that function as transcriptional activators or repressors.22,23 The best characterized gene fusions are in Ewing’s sarcoma (EWS-FLI1), clear cell sarcoma (EWS-ATF1), myxoid/round cell liposarcoma (TLS-CHOP), alveolar rhabdomyosarcoma (PAX3-FHKR), desmoplastic small round cell tumor (EWS-WT1), and synovial sarcoma (SS18-SSX). Fusion gene–related sarcomas have been estimated to account for 30% or more of all sarcomas.24
| DIAGNOSIS | CHROMOSOMAL ABNORMALITY | GENES INVOLVED |
|---|---|---|
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) t(1;13)(p36;q14) | PAX3-FKHR PAX7-FKHR |
| Alveolar soft part sarcoma | t(X;17)(p11.2;q25) | TFE3-ASPL |
| Angiomatoid fibrous histiocytoma | t(12;16)(q13;p11) | FUS-ATF1 |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWS-ATF1 |
| Congenital fibrosarcoma/congenital mesoblastic nephroma | t(12;15)(p13;q25) | ETV6-NTRK3 |
| Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | PDFGB-COL1A1 |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWS-WT1 |
| Endometrial stromal sarcoma | t(7;17)(p15;q21) | JAZF1-JJAZ1 |
| Ewing’s sarcoma/peripheral primitive neuroectodermal tumor | t(11;22)(q24;q12) t(21;22)(q22;q12) t(7;22)(p22;q12) t(17;22)(q12;q12) t(2;22)(q33;q12) t(16;21)(p11;q22) | EWS-FLI1 EWS-ERG EWS-ETV1 EWS-FEV EWS-E1AF FUS-ERG |
| Low-grade fibromyxoid sarcoma | t(7;16)(q33;p11) | FUS-CREB3I2 |
| Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) t(2;19)(p23;p13) t(2;17)(p23;q23) | TPM3-ALK TPM4-ALK CLTC-ALK |
| Myxoid liposarcoma | t(12;16)(q13;p11) t(12;22)(q13;q12) | TLS-CHOP EWS-CHOP |
| Myxoid chondrosarcoma | t(9;22)(q22;q12) t(9;15)(q22;q21) t(9;17)(q22;q11) | EWS-CHN TFC12-CHN TAF2N-CHN |
| Synovial sarcoma | t(x;18)(p11;q11) | SSX1-SYT SSX2-SYT SSX4-SYT |
Direct or indirect interactions between fusion transcripts and cell cycle regulators have been elucidated by several investigators and identify these transcripts as potentially promising molecular therapeutic targets.25 However, fusion genes in sarcoma have been successfully targeted in only a few cases, in which fusion resulted in overexpression of a growth factor or growth factor receptor. Several growth factors and their receptors (e.g., epidermal growth factor receptor) previously reported to play an important role in autocrine or paracrine stimulation of carcinoma growth have been associated with high histologic grade and poor prognosis in soft tissue sarcomas.
Oncogenes are genes that can induce malignant transformation and tend to drive cell proliferation. Several oncogenes have been associated with soft tissue sarcomas, including MDM2, N-myc, c-erbB2, and members of the ras family. These oncogenes produce specific oncoproteins that either play a role in nuclear function and cellular signal transduction or function as growth factors or growth factor receptors. This typically occurs in dedifferentiated liposarcoma, where the amplification of MDM2 drives the neoplastic process. Amplification of these genes has been shown to correlate with adverse outcome in several types of soft tissue sarcoma.22
GISTs are the classic example of sarcomas in which tumorigenesis is primarily driven by a single activating mutation, in the gene encoding KIT receptor tyrosine kinase or platelet-derived growth factor receptor-α (PDGFRA).19 The majority of GISTs have mutations in exon 11 of KIT and respond dramatically to the tyrosine kinase inhibitor imatinib mesylate, although this treatment rarely produces cure.
The largest group of sarcomas is the group with complex cytogenetic alterations, which includes high-grade spindle cell sarcomas and pleomorphic sarcomas.19 Many sarcomas in this group exhibit inactivation of tumor suppressor genes. The two genes most relevant to soft tissue sarcoma are retinoblastoma (Rb) and p53. Mutations or deletions in Rb can lead to retinoblastoma, the most common malignant ocular neoplasm of childhood. Survivors of retinoblastoma are at risk for developing soft tissue and bone sarcomas later in life. Patients with germline mutations in p53 (Li-Fraumeni syndrome) have a high incidence of soft tissue sarcomas Mutant p53 expression is thought to correlate with poor overall survival.22 Strategies to target p53 mutation are being investigated for treatment of some sarcomas.
Neurofibromatosis type 1 (von Recklinghausen’s disease) occurs in approximately 1 of every 3000 people and is due to various mutations in the NF-1 tumor suppressor gene, located on chromosome 17. Patients with neurofibromatosis type 1 have an estimated 3% to 15% additional risk of malignant disease compared with the general population lifetime risk, including malignant peripheral nerve sheath tumors (MPNST) and GIST. In turn, 25% to 50% of patients with MPNST have a mutation in NF-1.26
INITIAL ASSESSMENT
The clinical behavior of most soft tissue sarcomas is determined by anatomic location (depth in relation to the investing fascia), histologic subtype and grade of aggressiveness, and size. The dominant pattern of metastasis is hematogenous, primarily to the lungs. Lymph node metastasis is rare (affecting <5% of patients) except in a few histologic subtypes, including epithelioid sarcoma, pediatric rhabdomyosarcoma, clear cell sarcoma, angiosarcoma, and, more rarely, synovial sarcoma and myxofibrosarcoma,.27
Soft tissue sarcomas most commonly present as an asymptomatic mass. Extremity sarcomas may present as a deep venous thrombosis, particularly in patients without significant risk factors for thrombosis.28 Tumors in the distal extremities are generally smaller, whereas tumors in the proximal extremities and retroperitoneum can grow quite large before becoming apparent. Tumors often grow centrifugally and can compress surrounding normal structures. Infrequently, tumor impingement on bone or neurovascular bundles produces pain, edema, and swelling. Less frequently, tumors cause obstructive gastrointestinal symptoms or neurologic symptoms related to compression of lumbar or pelvic nerves. Often an extremity mass is discovered after a traumatic event that draws attention to a pre-existing lesion.
The differential diagnosis of a soft tissue mass should include consideration of lipoma (which is 100 times more common than sarcoma), lymphangioma, leiomyoma, neurinoma, primary or metastatic carcinoma, melanoma, and lymphoma. Superficial small lesions (<5 cm) that are new or that are not enlarging as indicated by clinical history can be observed. Enlarging masses and masses larger than 5 cm or deep to the fascia should be evaluated with a history, imaging, and biopsy.29
Diagnostic imaging should be performed before any invasive procedure to avoid the possibility of soft tissue swelling or hemorrhage complicating the image interpretation. Pretreatment diagnostic imaging is helpful for defining the size and anatomic location of a tumor and its proximity to adjacent structures; staging disease with respect to regional or metastatic spread; guiding percutaneous biopsy; and establishing whether a tumor is benign or malignant and low grade or high grade.
Radiographs are useful in the evaluation of primary bone tumors but not in the evaluation of soft tissue sarcomas of the extremities unless there is underlying bone involvement from an adjacent soft tissue tumor. Magnetic resonance imaging (MRI) is the preferred imaging technique for soft tissue sarcomas of the extremities, whereas computed tomography (CT) is most useful for evaluating retroperitoneal, intra-abdominal, and truncal sarcomas.30 CT of the chest should be performed to assess for lung metastases in patients with high-grade tumors larger than 5 cm. Abdominal/pelvic CT should be performed in patients with myxoid round cell liposarcomas, leiomyosarcomas, epithelioid sarcomas, or angiosarcomas because of their propensity to metastasize to the abdomen and/or pelvis.1 MRI of the brain should be considered for patients with alveolar soft part sarcomas and angiosarcomas because of their propensity to metastasize to the brain.
Ultrasonography may have a diagnostic role in patients with soft tissue sarcoma who cannot undergo MRI. Ultrasonography can also be a useful adjunct to MRI when findings on MRI are indeterminate and for delineating adjacent vascular structures. Finally, ultrasonography can be used for postoperative surveillance and to guide biopsies.
Chest CT should be performed to evaluate for lung metastasis at presentation and before any radical treatment. CT is also the preferred imaging technique for evaluating retroperitoneal sarcomas (Fig. 36-2).30 Current CT techniques can provide a detailed image of the abdomen and pelvis and can delineate adjacent organs and vascular structures. For extremity sarcomas, CT may be useful if MRI is not available or cannot be used. When histologic assessment of an extremity sarcoma reveals a myxoid liposarcoma, CT of the abdomen and pelvis should be done because this subtype is known to metastasize to the abdomen.31

MRI is the most useful imaging modality for extremity sarcomas because of its superior soft tissue contrast resolution and multiplanar capabilities. MRI accurately delineates muscle groups and distinguishes among bone, vascular structures, and tumor. Sagittal and coronal views allow evaluation of anatomic compartments in three dimensions (Fig. 36-3). Soft tissue sarcomas of the extremities usually present on MRI as a heterogeneous mass. Their signal intensity tends to be equal to or slightly higher than that of adjacent skeletal muscle on T1-weighted images and heterogeneous and high on T2-weighted images. Hemorrhagic, cystic, or necrotic changes may also be observed in the tumor. If adjacent vascular structures must be delineated, special MRI techniques may be performed, including magnetic resonance angiography. MRI may also be an important adjunct to cytologic analysis in distinguishing benign lesions such as lipomas, hemangiomas, schwannomas, neurofibromas, and intramuscular myxomas from their malignant counterparts. In patients undergoing preoperative chemotherapy, contrast-enhanced T1-weighted MRI can be useful in evaluating intratumoral necrosis.
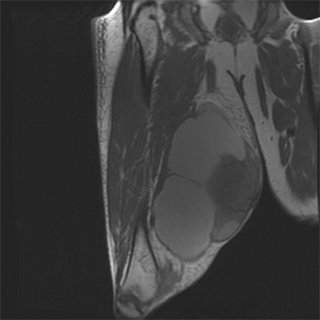
MRI is also valuable for assessing tumor recurrence after surgery. A baseline image is usually obtained 3 months after surgery. Some clinicians forego routine postoperative imaging of the primary extremity tumor site in asymptomatic patients, citing the difficulties in detecting early recurrence in scarred, irradiated tissue.30 Others advocate routine imaging every 3 to 4 months for the first 2 years, every 6 months in years 3 through 5, and then annually.
Positron emission tomography (PET) is a functional imaging modality that measures tumor uptake of the glucose analog [18F] fluorodeoxyglucose (FDG). PET imaging allows evaluation of the entire body. Although PET/CT may be useful in specific circumstances, FDG-PET is not currently recommended for the initial staging of patients with soft tissue sarcoma.
Roberge and colleagues compared FDG-PET/CT versus chest CT alone in the initial staging of 75 patients with soft tissue sarcoma and found that only one patient had disease upstaged as a result of PET, whereas two had false-positive findings and three had indeterminate findings with no subsequent development of metastasis.32 Previous studies that reported a marginal benefit of PET/CT for detecting metastasis at the time of sarcoma staging included patients with more heterogeneous tumors, such as osseous tumors, soft tissue osteosarcomas, Ewing’s sarcoma, and rhabdomyosarcoma.33,34,35
In patients with sarcoma, PET has primarily been used to assist with tumor grading and to assess response to chemotherapy.36,37,38,39 In 50 patients with resectable high-grade soft tissue tumors scheduled for preoperative chemotherapy and tumor resection, a 35% or greater reduction in tumor FDG uptake following an initial cycle of chemotherapy was associated with histopathologic tumor response defined as pathologic necrosis in 95% or more of the resected specimen.40
At centers where cytopathologists have experience with evaluation of mesenchymal tumors, fine-needle aspiration is an acceptable method of diagnosing most soft tissue sarcomas, particularly when the results correlate closely with clinical and radiologic findings.41 Fine-needle aspiration of primary tumors has a lower diagnostic accuracy rate (60%–90%) than core needle biopsy and is often not sufficient for establishing a specific histologic diagnosis and grade.42 However, fine-needle aspiration is the procedure of choice to confirm or rule out the presence of a metastatic focus or local recurrence.43
Although fine-needle aspiration of superficial lesions can often be done in the clinic, fine-needle aspiration of deeper tumors may need to be done by an interventional radiologist under sonographic or CT guidance. Generally, a 21- to 23-gauge needle is introduced into the mass after appropriate cleansing of the skin and injection of local anesthetic. Negative pressure is applied, and the needle is moved back and forth several times in various directions. After the negative pressure is released, the needle is withdrawn, and the contents of the needle are used to prepare smears.44 A cytopathologist then examines the slides to determine whether sufficient diagnostic material is present.
Core needle biopsy is safe, accurate,45,46 and economical47 and has become the preferred technique for diagnosing soft tissue lesions. Dupuy and colleagues found that core needle biopsy had an accuracy of 93% in 221 patients with musculoskeletal neoplasms.45
Sonography/CT guidance can prevent sampling of nondiagnostic necrotic or cystic areas of the tumor and thus increase the positive yield rate. Sonography/CT guidance also permits biopsy of tumors in otherwise inaccessible locations and tumors located near vital structures.
The tissue sample obtained from core needle biopsy is usually sufficient for several diagnostic tests, such as electron microscopy, cytogenetic analysis, and flow cytometry. The reported complication rate for core needle biopsy is less than 1%.45,46
Historically, an open surgical biopsy was the gold standard for achieving adequate tissue for definitive and specific histologic diagnosis of bone or soft tissue sarcomas. Contemporary guidelines recommend incisional biopsy when core needle biopsy cannot produce adequate tissue for diagnosis or when findings on core needle biopsy are nondiagnostic.
The disadvantages of incisional biopsy include the need to schedule the procedure, the need for general anesthesia, and high costs. In addition, an inappropriately placed incisions can necessitate more extensive definitive resection to encompass the biopsy site. In a series of 107 patients with soft tissue sarcoma, planned surgical treatments had to be changed because of poorly oriented biopsies in 25% of cases.48 Complication rates up to 17% have been reported after incisional biopsies.44 Potential complications include hematoma, infection, wound dehiscence, and tumor fungation, any of which can delay definitive treatment.44
Incisional biopsies should be performed only by surgeons experienced in the management of soft tissue sarcoma, ideally in a center specializing in the treatment of sarcoma and by the surgeon who will perform the definitive surgery. The biopsy incision should be oriented longitudinally along the extremity to allow a subsequent wide local excision that encompasses the biopsy site, scar, and tumor en bloc. A poorly oriented biopsy incision often necessitates an excessively large surgical defect for a wide local excision, which in turn can result in a larger postoperative radiation therapy field to encompass all tissues at risk. Adequate hemostasis must be achieved at the time of biopsy to prevent dissemination of tumor cells into adjacent tissue planes by hematoma.
Excisional biopsy can be performed for easily accessible (superficial) extremity or truncal lesions smaller than 3 cm. However, excisional biopsy rarely provides benefits over other biopsy techniques. Excisional biopsies should not be performed for lesions involving the hands and feet because such biopsies may complicate definitive re-excision. For sarcomas with initial diagnosis confirmed with excisional biopsy, microscopic residual disease has been reported in up to 69% of re-excision specimens49,50; without re-excision, the reported rate of local recurrence is 30% to 40% when margins are positive or uncertain.
Wide en bloc excision is seldom performed as a diagnostic procedure. When en bloc excision is done for diagnosis, the margin status is often not adequately evaluated during pathologic assessment of the specimen. Unless detailed descriptions of the surgical procedure and the pathology specimen are provided, the margins should be classified as uncertain or unknown, a classification associated with the same prognosis as resection margins that are positive for tumor cells. In patients with uncertain or unknown margins, re-excision should be performed if possible to ensure negative margins. The biopsy site or tract (if applicable) should be included en bloc with the re-resected specimen.
Sarcoma is generally diagnosed by morphologic assessment based on microscopic examination of histologic sections by an experienced sarcoma pathologist. However, even expert sarcoma pathologists disagree on the specific histologic diagnosis and the tumor grade in 25% to 40% of cases.51
Morphologic assessment can be supported by ancillary techniques, including conventional cytogenetics; immunohistochemistry; and molecular genetic testing, which is useful for classifying soft tissue sarcoma subtypes with multiple genetic aberrations. Other molecular diagnostic techniques include cytogenetic analysis, fluorescence in situ hybridization, and polymerase chain reaction–based methods.52 However, molecular genetic techniques are associated with significant technical limitations and should be interpreted in the context of the sarcoma’s morphologic features.
Some experts have suggested that pathologic classification of soft tissue sarcomas has more prognostic significance than does tumor grade when other pretreatment variables are taken into account. Tumors with limited metastatic potential include desmoid, atypical lipomatous tumor (also called well-differentiated liposarcoma), dermatofibrosarcoma protuberans, and solitary fibrous tumor. Tumors with an intermediate risk of metastatic spread usually have a large myxoid component and include myxoid liposarcoma, myxofibrosarcoma, and extraskeletal myxoid chondrosarcoma. Among the highly aggressive tumors with substantial metastatic potential are angiosarcoma, clear cell sarcoma, pleomorphic and dedifferentiated liposarcoma, leiomyosarcoma, MPNST, rhabdomyosarcoma, and synovial sarcoma.
It has recently been noted that malignant fibrous histiocytoma is not associated with a distinct gene cluster, suggesting that malignant fibrous histiocytoma is not a separate tumor entity but rather a common morphologic appearance of various sarcoma subtypes.53,54 For example, most tumors initially diagnosed as malignant fibrous histiocytoma in the retroperitoneum have been reclassified using genomic profiling as dedifferentiated liposarcomas,55 whereas those in the extremities have been reclassified as leiomyosarcoma, myxofibrosarcoma, or pleomorphic undifferentiated sarcoma.
Guidelines for the pathologic reporting of sarcoma have been established.1 Included in the report should be the primary diagnosis, anatomic site, depth, size, and histologic grade, presence or absence of necrosis, status of excision margins and lymph nodes, TNM stage, and additional features of the tumor (i.e., mitotic rate and presence or absence of vascular invasion).
Soft tissue sarcoma is most commonly staged using either the American Joint Committee on Cancer (AJCC) system (generally used in the United States) or the World Health Organization system. A unique aspect of sarcoma staging is the inclusion of tumor grade, which is one of the most important prognostic factors.56
The seventh edition of the AJCC staging system for soft tissue sarcomas is based on histologic grade of aggressiveness, tumor size and depth, and the presence of nodal or distant metastases.57 This system does not apply to GIST, fibromatosis (desmoid tumor), Kaposi’s sarcoma, or infantile fibrosarcoma.
Histologic grade is the most important prognostic factor for patients with soft tissue sarcoma. For accurate determination of grade, an adequate tissue sample must be appropriately fixed, stained, and reviewed by an experienced sarcoma pathologist. The features that define grade are cellularity, differentiation (good, moderate, or poor/anaplastic), pleomorphism, necrosis (absent, <50%, or ≥50%), and number of mitoses per high-power field (<10, 10–19, or ≥20). Tumor grade has been shown to predict metastasis and overall survival.58 Metastasis has been estimated to occur in 5% to 10% of low-grade lesions, 25% to 30% of intermediate-grade lesions, and 50% to 60% of high-grade lesions.
The number of grades varies according to the classification system used. The most common classification systems, those of the National Cancer Institute and the French Federation of Cancer Centers, use three-tier tumor grades.59 The National Cancer Institute system is based primarily on histologic subtype, location, and amount of necrosis. The French Federation of Cancer Centers system is based on tumor differentiation (good, moderate, or poor/anaplastic), number of mitoses per high-power field (<10, 10–19, or ≥20), and amount of tumor necrosis (absent, <50%, or ≥50%). A comparative analysis of the two systems suggested that the French Federation of Cancer Centers system has better prognostic capability, predicting 5-year survival rates of 90%, 70%, and 40% for grade 1, 2, and 3 tumors, respectively.59
Following the recommendation of the College of American Pathologists, the committee that developed the 2008 AJCC staging system changed the system from a four-grade to a three-grade system in which the grades are well differentiated (grade 1), moderately differentiated (grade 2), and poorly differentiated (grade 3).60 Grade 1 is considered low grade, and grades 2 and 3 are considered high grade.
Tumor size is an important prognostic variable in soft tissue sarcomas. Sarcomas have classically been stratified into two size groups; T1 lesions are 5 cm or smaller, and T2 lesions are larger than 5 cm. Some authors have suggested adding a third group for tumors larger than 15 cm, because such tumors are associated with a worse prognosis than tumors measuring between 5 and 15 cm.61,62
Anatomic tumor location was incorporated into the AJCC staging system in 1998. Soft tissue sarcomas above the superficial investing fascia of the extremity or trunk are designated “a” lesions within the T category, whereas tumors invading or deep to the fascia and all retroperitoneal, mediastinal, and visceral tumors are designated “b” lesions.
Overall, lymph node metastases arising from soft tissue sarcomas are rare,27 but the incidence of nodal involvement is higher for epithelioid sarcoma, pediatric rhabdomyosarcoma, clear cell sarcoma, synovial sarcoma, myxofibrosarcoma, and angiosarcoma. In the seventh edition of the AJCC staging system, sarcoma associated with nodal metastases was reclassified as stage III rather than stage IV because several studies reported better survival for patients with isolated regional lymph node metastases treated with radical lymphadenectomy than for patients with distant metastases.27,63,64,65 Patients with clinically or radiologically suspicious regional nodes should have metastases confirmed or ruled out by fine-needle aspiration before radical lymphadenectomy.
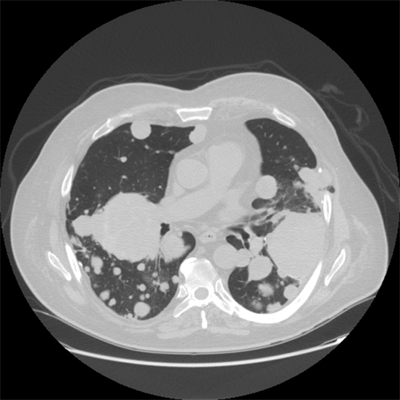
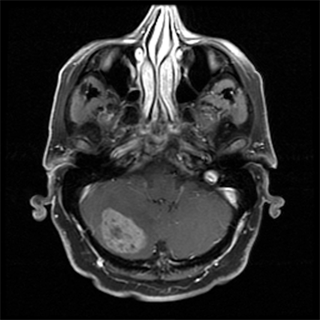
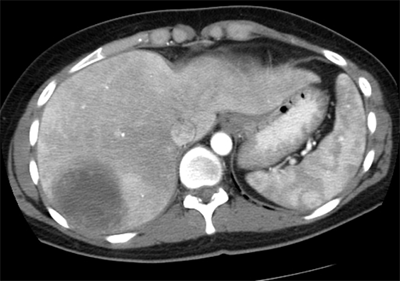
Distant metastases occur most often in the lungs (Fig. 36-4). Selected patients with pulmonary metastases may survive for long periods after surgical resection and chemotherapy. Other potential sites of metastasis include bone (Fig. 36-5), brain (Fig. 36-6), and liver (Fig. 36-7). Visceral and retroperitoneal sarcomas have a higher incidence of liver and peritoneal metastases.

Prognostic variables in soft tissue sarcoma include primary tumor size, grade, and depth, all of which are incorporated into the staging system, as well as histology, tumor site, and presentation (local recurrence or initial diagnosis). Patient factors such as older age and gender have also been associated with recurrence and mortality in several studies.66 A positive microscopic margin and early recurrence after resection of an extremity sarcoma have been shown to be associated with decreased survival.67
Several groups have reported that Ki-67, a proliferation marker, is correlated with a poor clinical outcome in high-grade extremity sarcomas.68,69 E-cadherin and catenins, proteins essential for intercellular junctions, have been associated with poor outcome in patients with soft tissue sarcoma.68 Similarly, higher CD100 expression has been shown to correlate with higher proliferative potential and poorer outcome.69
Prognostic nomograms for soft tissue sarcoma have been introduced for use in patient counseling, selecting appropriate surveillance strategies, and selecting patients for clinical trials.70 One such nomogram, developed by Kattan and colleagues at Memorial Sloan-Kettering Cancer Center, considers age, histology, grade, location, depth, and size to determine the likelihood of 12-year sarcoma-specific survival.70 Two validation studies using the nomogram demonstrated good predictive value.71 More recently, the same group of investigators developed histology subtype-specific nomograms for patients with liposarcoma, synovial sarcoma, and GIST72 and demonstrated that they were accurate in predicting disease-specific survival. Other investigators have just developed a site-specific nomogram for patients with retroperitoneal sarcoma, demonstrating an accurate prediction of survival and disease recurrence.73
TREATMENT OF EXTREMITY AND TRUNK WALL SARCOMA
The goals of treatment of soft tissue sarcoma are to maximize the likelihood of long-term recurrence-free survival while minimizing morbidity and maximizing function. In the past two decades, a multimodality treatment approach with optimal sequencing of treatments for individual patients has been shown to improve survival.74 Furthermore, patients with soft tissue sarcoma treated at high-volume centers have been shown to have improved survival and functional outcomes.75 Care at such centers is particularly important for patients with high-risk and advanced disease.
The overall 5-year survival rate for patients with all stages of soft tissue sarcoma is 50% to 60%. For patients with extremity sarcomas, a multidisciplinary treatment approach has resulted in local control rates exceeding 90% and 5-year survival rates exceeding 70%. Most patients who die of soft tissue sarcoma die of metastatic disease, which becomes evident within 2 to 3 years of initial diagnosis in 80% of cases.
Recommendations for evaluation and treatment of patients presenting with soft tissue masses are summarized in Table 36-3.
1. Soft tissue tumors that are enlarging or greater than 3 cm should be evaluated with radiologic imaging (ultrasonography or computed tomography [CT]), and a tissue diagnosis should be made using core needle biopsy. 2. Once a sarcoma diagnosis is established, obtain imaging (magnetic resonance imaging for extremity lesions and CT for other anatomic locations) and evaluate for metastatic disease with chest CT for intermediate- or high-grade (grade 2 or 3) or large (T2) tumors. 3. A wide local excision with 1- to 2-cm margins is adequate therapy for low-grade lesions and T1 tumors. 4. Radiation therapy plays a critical role in the management of large (T2), intermediate- or high-grade tumors. 5. Patients with locally advanced high-grade sarcomas or distant metastases should be evaluated for chemotherapy. 6. An aggressive surgical approach should be taken in the treatment of patients with an isolated local recurrence or resectable distant metastases. |
Primary tumors with no evidence of distant metastasis are managed with surgery alone or, when wide pathologic margins cannot be achieved because of anatomic constraints and/or the grade is high, surgery plus radiation therapy. The type of surgical resection is determined by several factors, including tumor location, tumor size, depth of invasion, involvement of nearby structures, need for skin grafting or autogenous tissue reconstruction, and the patient’s performance status. In 1985, the National Institutes of Health developed a consensus statement recommending limb-sparing surgery for most patients with high-grade extremity sarcomas.76 However, for patients with primary or recurrent tumors that cannot be grossly resected with a limb-sparing procedure and preservation of function (<5% of patients), amputation remains the treatment of choice.
Margin status after surgical resection has been shown to be an independent prognostic factor.77,78 The goal of surgical resection is to achieve a complete resection because microscopically positive or grossly positive resection margins are associated with increased risk of local recurrence and death.79 If an unexpected positive margin is found on pathologic examination of the resection specimen, re-excision should be performed. In patients with a positive margin, particularly in patients with macroscopic residual disease, local control is unlikely even with the addition of postoperative radiation therapy.80
The preferred treatment for extremity sarcomas is wide local excision that includes resection of the biopsy site. The goal of wide local excision is to remove the tumor with approximately 1 to 2 cm of surrounding normal soft tissue,77 but narrower margins may be necessary to preserve uninvolved critical neurovascular structures and may be adequate for patients undergoing radiation therapy.81 Dissection should proceed through grossly normal tissue planes not abutting the tumor. Soft tissue sarcomas are generally surrounded by a zone of compressed reactive tissue that forms a pseudocapsule, but this pseudocapsule should not be used to guide resection (enucleation). If the tumor is adjacent to or displacing major neurovascular structures, these do not need to be resected, but the adventitia or perineurium should be removed.1 For some massive tumors of the extremities, wide local excision entails a radical or complete anatomic compartment resection. Surgical clips should be placed to delineate the extent of the resection bed for patients likely to require postoperative radiation therapy.
Recent reports demonstrate encouraging results following radical en bloc resection with vascular reconstruction in the lower extremities.82,83 While en bloc resection with vascular reconstruction has been associated with increased rates of postoperative complications, reported local recurrence and 5-year survival rates are similar to those for patients not requiring vessel resection.84,85 Similarly, studies have shown acceptable functional outcomes with resection of the sciatic, tibial, and peroneal nerves with appropriate reconstruction and rehabilitation.86
Bone invasion from extremity soft tissue sarcoma, which can generally be identified using high-quality cross-sectional imaging such as MRI, has been estimated to occur in about 5% of patients and is associated with reduced overall survival.87 In cases of bone invasion, bone resection is required to obtain an adequate surgical margin and to achieve local control. Although tumor resection and repair of skeletal defects are possible, the likelihood of postoperative complications may be increased, and functional outcomes may be less favorable. Lin and colleagues88 recently analyzed 55 patients with soft tissue sarcomas abutting bone and reported that in the absence of frank cortical bone penetration, periosteum was an adequate surgical margin in patients treated with wide local excision and radiation.
Soft tissue sarcomas arising in the distal extremities, particularly the hands and feet, present unique technical challenges. While distal-extremity tumors are often detected at a smaller size (<5 cm) than proximal-extremity tumors, resection and reconstruction techniques are often more complex for distal-extremity tumors, and preoperative planning is critical to obtain favorable functional outcomes. Identifying the proximity of the tumor to underlying critical structures (e.g., bone, tendon, or neurovascular structures) using MRI is essential for surgical planning. In a reported series of patients with sarcomas of the hands or feet treated with limited surgery only, 32% of patients had local recurrences.89 Preservation of function and acceptable recurrence rates with limited surgery and adjuvant radiation therapy for soft tissue sarcomas of the distal extremities have been reported.90 For locally advanced tumors, repair of bone defects, vascular reconstruction, tendon transfers, and soft tissue reconstruction using regional or free flaps have resulted in good functional outcomes.91 Amputation remains a reasonable option for patients with soft tissue sarcomas of the distal extremities when acceptable oncologic or functional outcomes cannot be achieved using available limb salvage techniques.
In an interesting study conducted in Ontario and Quebec, investigators found patients expecting a difficult recovery and patients with uncertain expectations had worse functional outcomes than patients anticipating an easy recovery, indicating that preoperative education including consultation with rehabilitation services may optimize outcomes.92 Furthermore, all patients undergoing resection of extremity sarcomas should undergo physical therapy beginning immediately after surgery and continuing until maximum function is achieved.1
Several studies have reported improved survival for patients with isolated regional lymph node metastases treated with radical lymphadenectomy.27,63,64,65 Patients with clinically or radiologically suspicious regional nodes should have metastases confirmed before radical lymphadenectomy. At our institution, we perform ultrasound-guided fine-needle aspiration of lymph nodes in selected patients with suspicious clinical or radiologic findings. The utility of sentinel lymph node biopsy has remained controversial despite the recognition that several histologic subtypes of high-grade sarcoma are known to have a propensity for lymph node metastasis. However, there have been no prospective studies of the sensitivity and specificity of sentinel lymph node biopsy for such tumors.
Amputation is the treatment of choice for the 5% of patients with primary or recurrent extremity tumors whose tumors cannot be grossly resected with limb-sparing procedures and preservation of function. Historically, local excision of large, high-grade soft tissue sarcomas resulted in local failure rates of 50% to 70%, even when a margin of normal tissue around the tumor was excised; consequently, radical resection or amputation was recommended. Today, however, the addition of radiation therapy to less radical surgical resection has made limb salvage possible in most cases.
A comparison of amputation versus limb-sparing surgery followed by adjuvant radiation therapy performed by the National Cancer Institute between 1975 and 1981 demonstrated no significant difference between the two groups in local recurrence or overall survival rate.93 Potter and colleagues49 later reviewed the entire National Cancer Institute experience with 123 patients treated with conservative surgery plus radiation therapy and 83 treated with amputation. The local recurrence rate was significantly higher in the surgery and adjuvant radiation therapy group: 8% versus 0% in the amputation group. However, survival rates did not differ between the groups. Several large single-institution studies have since also reported favorable local control rates with conservative resection plus radiation therapy.94,95,96
Isolated regional perfusion is a limb-sparing technique in which a soft tissue sarcoma is perfused with high concentrations of tumor necrosis factor-α and melphalan under hyperthermic conditions. The technique is generally used for locally advanced, multifocal, or locally recurrent disease; it has also served as a palliative treatment to achieve local control for patients with distant metastases.
Limb perfusion requires isolating the main artery and vein of the perfused limb from the systemic circulation. The anatomic approach is determined by tumor site: external iliac vessels are used for thigh tumors, femoral or popliteal vessels for calf tumors, and axillary vessels for upper extremity tumors. The vessels are dissected, and all collateral vessels are ligated. The main artery and vein are then cannulated and connected to a pump oxygenator similar to that used in cardiopulmonary bypass. Either a tourniquet or an Esmarch band is applied to the limb to achieve complete vascular isolation. Chemotherapeutic agents are then added to the perfusion circuit and circulated for 90 minutes. Systemic leakage from the perfused limb is monitored continuously with 99Tc-radiolabeled human serum albumin injected into the perfusate, and radioactivity above the precordial area is recorded with a Geiger counter. During the entire procedure, hyperthermia of the perfused limb is maintained by external heating and by warming the perfusate to 40°C. At the end of the procedure, the limb is washed out, the cannulas are extracted, and the blood vessels are repaired.
Despite the 40-year history of using isolated limb perfusion to treat extremity sarcomas, many questions about this technique remain to be answered. The optimal chemotherapeutic agent in the perfusion circuit, the benefits of hyperthermia, and the effectiveness of hyperthermic perfusion as neoadjuvant or adjuvant treatment remain to be elucidated. Studies published to date have involved heterogeneous patient groups and various chemotherapeutic agents. Despite these limitations, response rates from 18% to 80% and overall 5-year survival rates from 50% to 70% have been reported.97,98,99,100,101 However, survival outcomes following isolated limb perfusion have not yet been directly compared with survival outcomes after more conventional treatment approaches.
In the initial report of isolated regional perfusion for extremity sarcomas, published in 1974, McBride reported results in 79 patients with extremity sarcomas who had been treated with isolated limb perfusion during the previous 14 years.97 All patients received melphalan and dactinomycin. The overall 5-year survival rate was 57%, and only 13 patients had subsequent amputation for recurrent disease. Over the next 20 years, isolated perfusion for treatment of extremity sarcoma fell out of favor for several reasons. Most notably, improved survival and decreased local recurrence rates could be obtained with less radical therapy, including conservative surgical excision combined with radiation to allow limb sparing in patients who were previously thought to require amputation.
A 1992 report by Lienard and colleagues101 renewed interest in isolated limb perfusion for extremity tumors. Those investigators reported a 100% response rate among patients with extremity melanomas and sarcomas treated with high-dose recombinant tumor necrosis factor-α plus interferon-γ and melphalan in an isolated perfusion circuit. This report led to larger studies geared specifically to patients with sarcoma. The largest of these studies, the European Multicenter Study, was reported by Eggermont and colleagues in 1996.99 In that study of 186 patients, the overall tumor response rate was 82%, and the clinical and pathologic complete response rate was 29%. Although all of the study participants were reported to initially be candidates for amputation, the rate of limb salvage following isolated limb perfusion was 82%.99 Subsequent studies have shown high local response and limb salvage rates and acceptable local and systemic toxic effects.102
However, results in the United States have been inferior to those reported in Europe. In a study by Fraker and colleagues, the complete response rate was 26%, and an additional 30% of patients had a partial response. Fourteen patients (32%) underwent amputation for progressive tumors, while the remaining 30 patients (68%) were able to undergo limb-sparing surgery after isolated limb perfusion.100 The inferior results in the U.S.-based studies are thought to be due to patient selection biases and the degree of treatment before limb perfusion.
Radiation therapy is part of the standard treatment for high-grade extremity and trunk wall soft tissue sarcomas either in the pre- or postoperative setting. Patients with low-grade tumors or small, superficial high-grade tumors that have been resected with adequate margins may safely avoid radiation therapy.
The evidence supporting adjuvant radiation therapy for patients eligible for conservative surgical resection comes from two randomized trials103,104 and three large single-institution reports.105,106,107 In a randomized trial by the National Cancer Institute, 91 patients with high-grade extremity tumors were treated with limb-sparing surgery followed by chemotherapy alone or radiation therapy plus chemotherapy. The 10-year local control rate was 98% for patients receiving radiation therapy compared with 70% for those not receiving radiation therapy (P = .0001).103 Similarly, in a randomized trial from Memorial Sloan-Kettering Cancer Center, 164 patients underwent conservative surgery followed by observation or brachytherapy. For patients with high-grade tumors, the 5-year local control rate was 66% in the observation group and 89% in the brachytherapy group (P = .003).104 For patients with low-grade tumors, no significant difference was observed between treatment groups.108
Until recently, the standard treatment guidelines required radiation therapy after surgery for all patients with intermediate- or high-grade tumors of any size. However, small tumors (≤5 cm) have not generally been associated with local recurrence, and radiation therapy for such tumors may not be necessary.104 In a series of 174 patients reported by Geer and colleagues, postoperative radiation therapy did not improve 5-year local recurrence or overall survival rates for patients with small soft tissue sarcomas.109 Karakousis and colleagues reported a 5-year local recurrence rate of 6% for 80 patients with extremity sarcomas treated with wide local excision and observation, a rate similar to that for the 64 patients who underwent resection with narrower surgical margins and postoperative radiation therapy.110
The optimal mode of radiation therapy (external-beam radiation therapy, brachytherapy, or intensity-modulated radiation therapy [IMRT]) and timing of radiation therapy (preoperative, intraoperative, or postoperative) have yet to be defined. External-beam radiation therapy can be delivered using photons or particle beams (electrons, protons, pions, or neutrons). Conventional fractionation is usually 1.8 to 2 Gy per day. CT is an integral part of radiation therapy, used to define the gross tumor volume and to estimate the margin of tissue at risk for microscopic tumor involvement. The optimal radiation margin is not well defined: a margin of 5 to 7 cm is standard, but some centers advocate wider margins for tumors larger than 15 cm. At most institutions, the typical preoperative dose is 50 Gy given in 25 fractions, and resection is performed 4 to 8 weeks after completion of radiation therapy to allow acute radiation changes to subside. Postoperative radiation therapy planning is based on tumor site, tumor grade, surgical margins, and institutional preferences. The entire surgical scar and drain sites should be included in the field so that a near-full dose can be administered to the superficial skin. Metallic clips placed in the tumor bed during surgery can help define the limits of the resection and aid in radiation therapy planning. Doses of 60 to 70 Gy are usually necessary for postoperative treatment.
No consensus exists on the optimal sequence of radiation therapy and surgery. The available data come largely from single-institution, nonrandomized studies. Proponents of preoperative radiation therapy note that multidisciplinary planning with radiation oncologists, medical oncologists, and surgeons is easier early in the course of therapy. In addition, for some radiosensitive histologic subtypes, such as myxoid liposarcoma, preoperative radiation therapy may shrink the tumor, facilitating resection with negative margins. Furthermore, a tissue bed undisturbed by resection has better tissue oxygenation and can be successfully treated with lower doses of radiation. In addition, Nielsen and colleagues111 demonstrated that preoperative radiation fields are smaller than postoperative radiation fields and that the average number of joints included in the field is lower with preoperative than postoperative radiation therapy, which may result in improved functional outcome. Critics of preoperative radiation therapy cite the difficulty of pathologic assessment of margins and the increased rate of postoperative wound complications.112 However, reconstructive surgical techniques with advanced tissue transfer procedures are being used more often in these high-risk wounds and reportedly result in better outcomes. The higher doses generally required for postoperative radiation therapy have also been shown to be associated with greater long-term functional impairment.
The only randomized comparison of preoperative and postoperative radiation therapy to date was performed by the National Cancer Institute of Canada Clinical Trials–Canadian Sarcoma Group.113
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


