|
Typical Clinical Features |
Microscopic Features |
Ancillary Investigations |
Alveolar rhabdomyosarcoma |
Adolescents and young adults
Rapidly growing mass
Extremities, head and neck, trunk |
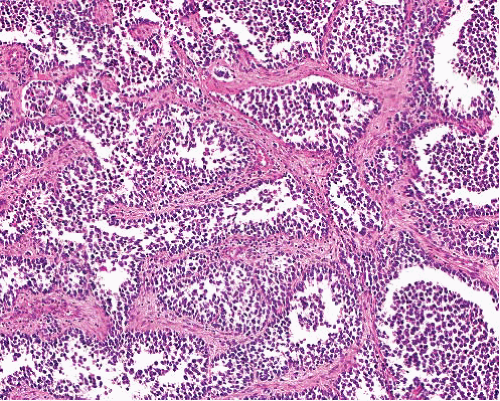
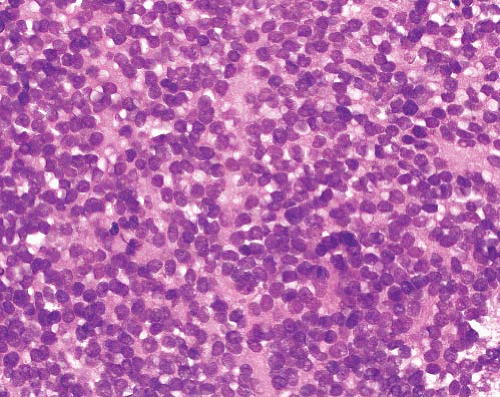
Sheets and nests of cells with round nuclei of small to intermediate size and scant cytoplasm, separated by fibrous septa
Central necrosis and peripheral cellular preservation causing classic “alveolar” morphology
Solid variant lacks alveolar pattern
Rhabdomyoblasts in some cases |
Desmin+, nuclear myogenin, and MyoD1+
Fluorescence in situ hybridization for FOXO1 rearrangements and RT-PCR for PAX3/7-FOXO1 fusion transcripts |
Ewing sarcoma/primitive neuroectodermal tumor |
Children and young adults
Extraskeletally in extremity deep soft tissue, paravertebral sites, retroperitoneum, thoracopulmonary region |
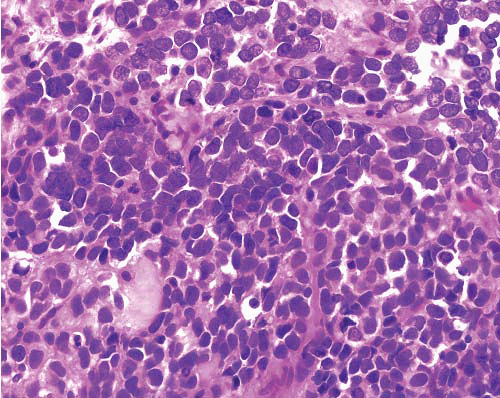
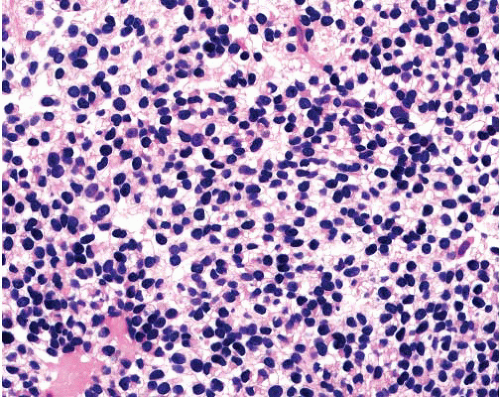
Sheets of uniform cells with round nuclei, fine chromatin and scant cytoplasm
Sometimes rosette formation |
Many tumors CD99+ (supporting diagnosis)
Recurrent chromosomal translocations resulting in EWS-ETS family fusion oncoproteins, detectable by reverse transcriptionpolymerase chain reaction
EWSR1 rearrangements detectable by fluorescence in situ hybridization
Minimal or absent intercellular reticulin |
CIC-DUX4-associated small round cell sarcoma |
Young adult males, but there is a wide age distribution (from first to seventh decade)
50% arise in limbs |
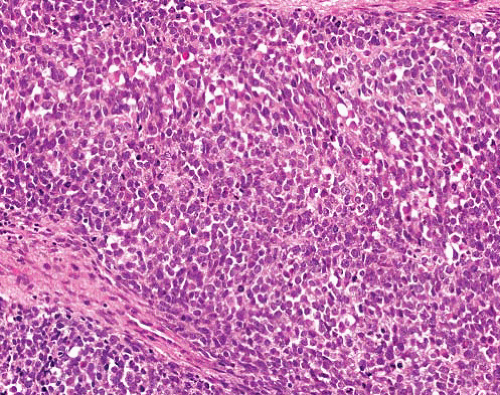
Sheets of small- to mediumsized round to oval cells, often prominent nucleoli, high mitotic activity and necrosis
Sometime spindling
Associated with a higher degree of morphologic heterogeneity than classical Ewing sarcoma |
Variable membranous CD99 (varying from diffuse to focal and weak)
WT1+, FLI variable
Very rarely express focal cytokeratin, EMA, desmin and S100 protein
CIC-DUX4 fusion (fluorescence in situ hybridization for CIC rearrangement) |
BCOR-CCNB3-associated round cell sarcoma |
Predominantly in bone; smaller numbers in deep soft tissues
Adolescents and young adults
Male preponderance |
Relatively wide histomorphologic spectrum; most predominantly showing undifferentiated, small round cell Ewing sarcoma-like morphology
Smaller numbers show spindle cell morphology |
CCNB3 immunohistochemostry
Also bcl-2+ and sometimes CD99 and CD117+
BCOR fluorescence in situ hybridization assay |
Poorly differentiated synovial sarcoma |
Young adults
Any site, but most common in extremity deep soft tissue, especially around knee and adjacent to joints and tendons |
Sheets of rounded to ovoid cells
More spindled cells may be seen focally
Mast cells |
CK, EMA, CD99, S100 protein all focally+, bcl-2+, TLE1+ (nuclear), CD34 and CD117−. Nested reticulin pattern t(X;18)(p11;q11), SS18-SSX fusion genes |
Extraskeletal mesenchymal chondrosarcoma |
Young adults
Head and neck, extremities
Exclude metastasis from extraosseous site |
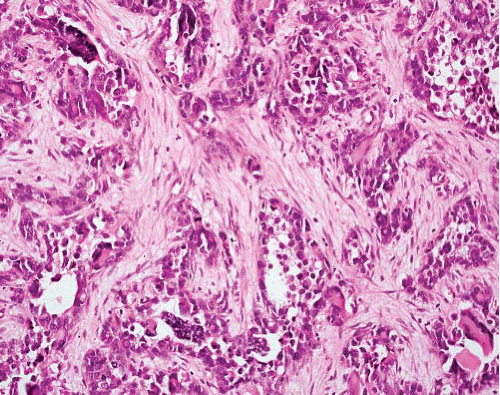
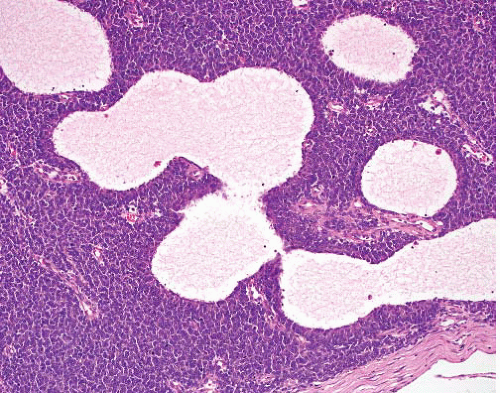
Biphasic morphology
Hypercellular sheets of primitive small cells with round, ovoid, or spindled nuclei
Interspersed islands of well-differentiated hyaline cartilage
Hemangiopericytic vascular pattern |
Small cell component CD99+
Cartilaginous component S100 protein+ |
Small cell osteosarcoma |
All ages, but most frequently in second decade |
Sheets of small, uniform, round to focally short spindled cells
Foci of malignant osteoid, and sometimes malignant cartilage |
Variable focal S100 protein+
CD99− |
Desmoplastic small round cell tumor |
Adolescents and young adults, particularly males
Abdomen or pelvis
Often widespread serosal involvement |
Sheets of tightly packed monotonous cells with small nuclei and usually minimal cytoplasm
Striking desmoplastic stromal reaction which divides the tumor into sharply demarcated islands
Necrosis common |
Nuclear expression of WT1 (carboxy terminal)
Divergent differentiation with coexpression of desmin, cytokeratins and neural markers t(11;22)(p13;q12) and EWSR1-WT1 fusion gene |
Neuroblastoma |
Infants and children in first 2 y of life
Follow distribution of sympathetic ganglia, with majority arising in retroperitoneum (of which about half arise in adrenal gland) |
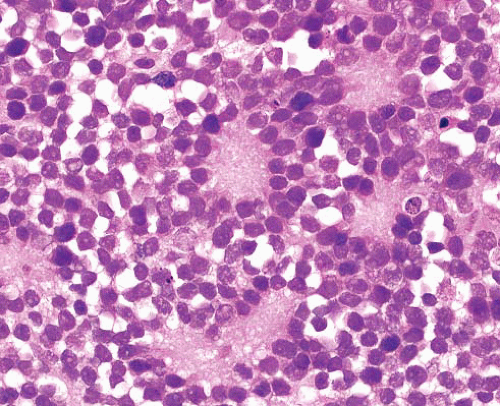
Sheets and lobules of small- to intermediate-sized cells, with hyperchromatic round nuclei
Pale neurofibrillary matrix resembling neuropil
Cells may show rosette formation
May show small amounts of ganglionic differentiation |
NSE+
Neurofilament+, NB84+
CD99−
Amplification of MYCN oncogene in approximately 25% of cases |
Ganglioneuroblastoma |
Retroperitoneum, mediastinum |
Neuroblastoma-like areas of primitive neuroblasts with areas of ganglioneuromatous stroma, subclassified according to arrangement and proportion of these areas |
Ganglion cells NSE+, CD56+, NF+
Spindle Schwann cells S100 protein+ |
Olfactory neuroblastoma |
Adults with bimodal peak (second and sixth decades)
Roof of nasal fossa, maxillary and ethmoid sinuses, nasopharynx |
Lobules and nests of uniform small cells with round vesicular nuclei and scanty cytoplasm, often within neurofibrillary background
Rosettes may be present |
NSE+, NFP+ variably, synaptophysin+, chromogranin+
S100 protein+ in sustentacularlike cells that surround islands of tumor
CD99− |
Ectomesenchymoma |
Children, M > F
Intracranial, nasal cavity, genitourinary tract, and soft tissue |
Neuroectodermal elements (usually ganglioneuroma or ganglioneuroblastoma) and mesenchymal component, usually rhabdomyosarcoma |
Appropriate immunoreactivity for each component |
Small cell malignant peripheral nerve sheath tumor |
Deep soft tissue of trunk and limbs |
Uniform, small, round cells with scanty cytoplasm
Sometimes foci of uniform, short, spindle-shaped tumor cells arranged in whorl-like patterns |
S100 protein+ focally, occasional GFAP+
CD99− |
Small cell carcinoma |
Older adults
Usually lung, but can occur in many other primary sites |
Sheets of small to medium round to fusiform cells with hyperchromatic nuclei
Nuclear molding, necrosis and frequent mitoses |
CK+ (dot), many TTF1+, chromogranin+, synaptophysin+, CK20−, NF− |
Merkel cell carcinoma |
Older adults
Face and extremities |
Tumor in dermis or subcutis
Monotonous round cells, multiple nucleoli |
Low molecular weight CK+, CK20+ in perinuclear dotlike distributions
NSE, neurofilament+
TTF1− |
Melanoma |
Primary dermal lesion may be apparent |
May have junctional component
Often in nested pattern
Melanin pigment |
S100 protein+, HMB45+, melan-A+ |
Lymphoma |
May have lymphadenopathy |
Cells have dispersed morphology, and tumor can have a more heterogeneous appearance with a mixed population of other inflammatory cells |
CD45+, CD20, CD3+, TdT+ (lymphoblastic lymphoma) |
Granulocytic sarcoma |
Any site, including skin and lymph nodes
Patient may have underlying acute myeloid leukemia |
Extramedullary tumor comprising immature myeloid (granulocytic or monocytic) cells |
Lysozyme, CD43+, variably myeloperoxidase, CD117, CD13, CD33+ |
Round cell liposarcoma |
Deep soft tissues of extremities of young adults
Often metastasizes to other soft tissue sites as well as to lungs |
Monotonous small uniform rounded cells with minimal myxoid matrix, with some lipoblasts
Rich network of delicate vessels becomes inconspicuous in round cell liposarcoma |
S100 protein+ in some cases
t(12;16)(q13;p11) or t(12;22) (q13;q12) with FUS-DDIT3 or EWSR1-DDIT3 fusion genes |
Endometrial stromal sarcoma |
Females
Can arise in a focus of endometriosis or present as metastasis in abdomen or elsewhere |
Short closely packed spindle cells, variable focal myoid differentiation
Thick-walled vessels |
CD10+, SMA+, occasional des+, CK+
Nuclear beta-catenin+ in 50% ER+, PgR + JAZF1-JJAZ1, JAZF1-PHF1, and EPC1-PHF1 fusion genes |
NUT midline carcinoma |
Mediastinum, head and neck
Occur over a wide age range
Roughly equal sex distribution |
Relatively uniform, undifferentiated round or epithelioid cells in sheets, or sometimes nests within desmoplastic stroma
Variable squamous differentiation |
Most are variably positive for p63, cytokeratins including CK7, and antibodies to the NUT protein
>50% show variable CD34+ CD99−; muscle, neural, melanocytic and hematolymphoid marker
Characterized by chromosomal rearrangements involving the gene encoding the nuclear protein of the testis (NUT) at 15q14
Most commonly due to t(15;19), fusing NUT with the BRD4 gene on chromosome 19, but in approximately a third of cases NUT is fused to a different partner gene |