KEY POINTS
Shock is defined as a failure to meet the metabolic demands of cells and tissues and the consequences that ensue.
A central component of shock is decreased tissue perfusion. This may be a direct consequence of the etiology of shock, such as in hypovolemic/hemorrhagic, cardiogenic, or neurogenic etiologies, or may be secondary to elaborated or released molecules or cellular products that result in endothelial/cellular activation, such as in septic shock or traumatic shock.
Physiologic responses to shock are based on a series of afferent (sensing) signals and efferent responses that include neuroendocrine, metabolic, and immune/inflammatory signaling.
The mainstay of treatment of hemorrhagic/hypovolemic shock includes volume resuscitation with blood products. In the case of hemorrhagic shock, timely control of bleeding is essential and influences outcome.
Prevention of hypothermia, acidemia, and coagulopathy is essential in the management of patients in hemorrhagic shock.
The mainstay of treatment of septic shock is fluid resuscitation, initiation of appropriate antibiotic therapy, and control of the source of infection. This includes drainage of infected fluid collections, removal of infected foreign bodies, and débridement of devitalized tissues.
A combination of physiologic parameters and markers of organ perfusion/tissue oxygenation are used to determine if patients are in shock and to follow the efficacy of resuscitation.
EVOLUTION IN UNDERSTANDING SHOCK
Shock, at its most rudimentary definition and regardless of the etiology, is the failure to meet the metabolic needs of the cell and the consequences that ensue. The initial cellular injury that occurs is reversible; however, the injury will become irreversible if tissue perfusion is prolonged or severe enough such that, at the cellular level, compensation is no longer possible. Our evolution in the understanding of shock and the disease processes that result in shock made its most significant advances throughout the twentieth century as our appreciation for the physiology and pathophysiology of shock matured. Most notably, this includes the sympathetic and neuroendocrine stress responses on the cardiovascular system. The clinical manifestations of these physiologic responses are most often what lead practitioners to the diagnosis of shock as well as guide the management of patients in shock. However, hemodynamic parameters such as blood pressure and heart rate are relatively insensitive measures of shock, and additional considerations must be used to help aid in early diagnosis and treatment of patients in shock. The general approach to the management of patients in shock has been empiric: assuring a secure airway with adequate ventilation, control of hemorrhage in the bleeding patient, and restoration of vascular volume and tissue perfusion.
Integral to our understanding of shock is the appreciation that our bodies attempt to maintain a state of homeostasis. Claude Bernard suggested in the mid-nineteenth century that the organism attempts to maintain constancy in the internal environment against external forces that attempt to disrupt the milieu interieur.2 Walter B. Cannon carried Bernard’s observations further and introduced the term homeostasis, emphasizing that an organism’s ability to survive was related to maintenance of homeostasis.3 The failure of physiologic systems to buffer the organism against external forces results in organ and cellular dysfunction, what is clinically recognized as shock. He first described the “fight or flight response,” generated by elevated levels of catecholamines in the bloodstream. Cannon’s observations on the battlefields of World War I led him to propose that the initiation of shock was due to a disturbance of the nervous system that resulted in vasodilation and hypotension. He proposed that secondary shock, with its attendant capillary permeability leak, was caused by a “toxic factor” released from the tissues.
In a series of critical experiments, Alfred Blalock documented that the shock state in hemorrhage was associated with reduced cardiac output due to volume loss, not a “toxic factor.”4 In 1934, Blalock proposed four categories of shock: hypovolemic, vasogenic, cardiogenic, and neurogenic. Hypovolemic shock, the most common type, results from loss of circulating blood volume. This may result from loss of whole blood (hemorrhagic shock), plasma, interstitial fluid (bowel obstruction), or a combination. Vasogenic shock results from decreased resistance within capacitance vessels, usually seen in sepsis. Neurogenic shock is a form of vasogenic shock in which spinal cord injury or spinal anesthesia causes vasodilation due to acute loss of sympathetic vascular tone. Cardiogenic shock results from failure of the heart as a pump, as in arrhythmias or acute myocardial infarction (MI).
This categorization of shock based on etiology persists today (Table 5-1). In recent clinical practice, further classification has described six types of shock: hypovolemic, septic (vasodilatory), neurogenic, cardiogenic, obstructive, and traumatic shock. Obstructive shock is a form of cardiogenic shock that results from mechanical impediment to circulation leading to depressed cardiac output rather than primary cardiac failure. This includes etiologies such as pulmonary embolism or tension pneumothorax. In traumatic shock, soft tissue and bony injury leads to the activation of inflammatory cells and the release of circulating factors, such as cytokines and intracellular molecules that modulate the immune response. Recent investigations have revealed that the inflammatory mediators released in response to tissue injury (damage-associated molecular patterns [DAMPs]) are recognized by many of the same cellular receptors (pattern recognition receptors [PRRs]) and activate similar signaling pathways as do bacterial products elaborated in sepsis (pathogen-associated molecular patterns), such as lipopolysaccharide.5 These effects of tissue injury are combined with the effects of hemorrhage, creating a more complex and amplified deviation from homeostasis.
In the mid to later twentieth century, the further development of experimental models contributed significantly to the understanding of the pathophysiology of shock. In 1947, Wiggers developed a sustainable, irreversible model of hemorrhagic shock based on uptake of shed blood into a reservoir to maintain a set level of hypotension.6 G. Tom Shires added further understanding of hemorrhagic shock with a series of clinical studies demonstrating that a large extracellular fluid deficit, greater than could be attributed to vascular refilling alone, occurred in severe hemorrhagic shock.7,8 The phenomenon of fluid redistribution after major trauma involving blood loss was termed third spacing and described the translocation of intravascular volume into the peritoneum, bowel, burned tissues, or crush injury sites. These seminal studies form the scientific basis for the current treatment of hemorrhagic shock with red blood cells and lactated Ringer’s solution or isotonic saline.
As resuscitation strategies evolved and patients survived the initial consequences of hemorrhage, new challenges of sustained shock became apparent. During the Vietnam War, aggressive fluid resuscitation with red blood cells and crystalloid solution or plasma resulted in survival of patients who previously would have succumbed to hemorrhagic shock. Renal failure became a less frequent clinical problem; however, a new disease process, acute fulminant pulmonary failure, appeared as an early cause of death after seemingly successful surgery to control hemorrhage. Initially called DaNang lung or shock lung, the clinical problem became recognized as acute respiratory distress syndrome (ARDS). This led to new methods of prolonged mechanical ventilation. Our current concept of ARDS is a component in the spectrum of multiple organ system failure.
Studies and clinical observations over the past two decades have extended the early observations of Canon, that “restoration of blood pressure prior to control of active bleeding may result in loss of blood that is sorely needed,” and challenged the appropriate endpoints in resuscitation of uncontrolled hemorrhage.9 Core principles in the management of the critically ill or injured patient include: (a) definitive control of the airway must be secured, (b) control of active hemorrhage must occur promptly (delay in control of bleeding increases mortality, and recent battlefield data would suggest that in the young and otherwise healthy population commonly injured in combat, control of bleeding is the paramount priority), (c) volume resuscitation with blood products (red blood cells, plasma, and platelets) with limited volume of crystalloid must occur while operative control of bleeding is achieved, (d) unrecognized or inadequately corrected hypoperfusion increases morbidity and mortality (i.e., inadequate resuscitation results in avoidable early deaths from shock), and (e) excessive fluid resuscitation may exacerbate bleeding (i.e., uncontrolled resuscitation is harmful). Thus both inadequate and uncontrolled volume resuscitation is harmful.
A modern definition and approach to shock acknowledges that shock consists of inadequate tissue perfusion marked by decreased delivery of required metabolic substrates and inadequate removal of cellular waste products.This involves failure of oxidative metabolism that can involve defects of oxygen (O2) delivery, transport, and/or utilization. Current challenges include moving beyond fluid resuscitation based on endpoints of tissue oxygenation, and using therapeutic strategies at the cellular and molecular level. This approach will help to identify compensated patients or patients early in the course of their disease, initiate appropriate treatment, and allow for continued evaluation for the efficacy of resuscitation and adjuncts.
Current investigations focus on determining the cellular events that often occur in parallel to result in organ dysfunction, shock irreversibility, and death. This chapter will review our current understanding of the pathophysiology and cellular responses of shock states. Current and experimental diagnostic and therapeutic modalities for the different categories of shock are reviewed, with a focus on hemorrhagic/hypovolemic shock and septic shock.
PATHOPHYSIOLOGY OF SHOCK
Regardless of etiology, the initial physiologic responses in shock are driven by tissue hypoperfusion and the developing cellular energy deficit.This imbalance between cellular supply and demand leads to neuroendocrine and inflammatory responses, the magnitude of which is usually proportional to the degree and duration of shock. The specific responses will differ based on the etiology of shock, as certain physiologic responses may be limited by the inciting pathology. For example, the cardiovascular response driven by the sympathetic nervous system is markedly blunted in neurogenic or septic shock. Additionally, decreased perfusion may occur as a consequence of cellular activation and dysfunction, such as in septic shock and to a lesser extent traumatic shock (Fig. 5-1). Many of the organ-specific responses are aimed at maintaining perfusion in the cerebral and coronary circulation. These are regulated at multiple levels including (a) stretch receptors and baroreceptors in the heart and vasculature (carotid sinus and aortic arch), (b) chemoreceptors, (c) cerebral ischemia responses, (d) release of endogenous vasoconstrictors, (e) shifting of fluid into the intravascular space, and (f) renal reabsorption and conservation of salt and water.
Figure 5-1.
Pathways leading to decreased tissue perfusion and shock. Decreased tissue perfusion can result directly from hemorrhage/hypovolemia, cardiac failure, or neurologic injury. Decreased tissue perfusion and cellular injury can then result in immune and inflammatory responses. Alternatively, elaboration of microbial products during infection or release of endogenous cellular products from tissue injury can result in cellular activation to subsequently influence tissue perfusion and the development of shock. HMGB1 = high mobility group box 1; LPS = lipopolysaccharide; RAGE = receptor for advanced glycation end products.
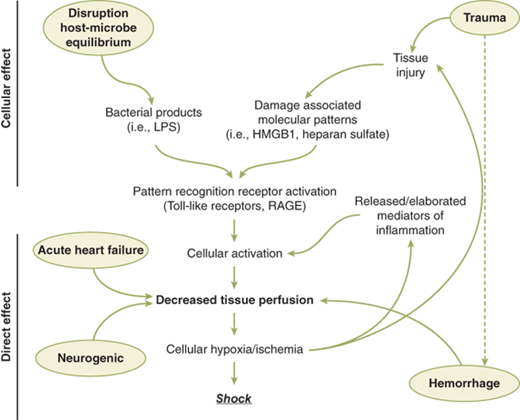
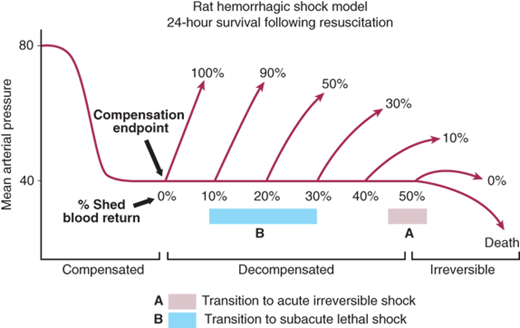
Furthermore, the pathophysiologic responses vary with time and in response to resuscitation. In hemorrhagic shock, the body can compensate for the initial loss of blood volume primarily through the neuroendocrine response to maintain hemodynamics. This represents the compensated phase of shock. With continued hypoperfusion, which may be unrecognized, cellular death and injury are ongoing and the decompensation phase of shock ensues. Microcirculatory dysfunction, parenchymal tissue damage, and inflammatory cell activation can perpetuate hypoperfusion. Ischemia/reperfusion injury will often exacerbate the initial insult. These effects at the cellular level, if untreated, will lead to compromise of function at the organ system level, thus leading to the “vicious cycle” of shock (Fig. 5-2). Persistent hypoperfusion results in further hemodynamic derangements and cardiovascular collapse. This has been termed the irreversible phase of shock and can develop quite insidiously and may only be obvious in retrospect. At this point, there has occurred extensive enough parenchymal and microvascular injury such that volume resuscitation fails to reverse the process, leading to death of the patient. In experimental animal models of hemorrhagic shock (modified Wiggers model), this is represented by the “uptake phase” or “compensation endpoint” when shed blood must be returned to the animal to sustain the hypotension at the set level to prevent further hypotension and death.10 If shed blood volume is slowly returned to maintain the set level of hypotension, eventually the injury progresses to irreversible shock, where further volume will not reverse the process and the animal dies (Fig. 5-3).
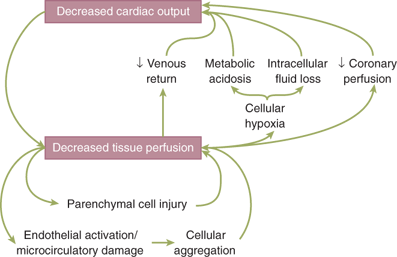
Figure 5-3.
Rat model of hemorrhagic shock through the phases of compensation, decompensation, and irreversibility. The percentages shown above the curve represent survival rates. (Adapted with permission from Lippincott Williams & Wilkins/Wolters Kluwer Health: Shah NS, Kelly E, Billiar TR, et al. Utility of clinical parameters of tissue oxygenation in a quantitative model of irreversible hemorrhagic shock. Shock. 1998;10:343-346. Copyright © 1998.)
The goal of the neuroendocrine response to hemorrhage is to maintain perfusion to the heart and the brain, even at the expense of other organ systems. Peripheral vasoconstriction occurs, and fluid excretion is inhibited. The mechanisms include autonomic control of peripheral vascular tone and cardiac contractility, hormonal response to stress and volume depletion, and local microcirculatory mechanisms that are organ specific and regulate regional blood flow. The initial stimulus is loss of circulating blood volume in hemorrhagic shock. The magnitude of the neuroendocrine response is based on both the volume of blood lost and the rate at which it is lost.
Afferent impulses transmitted from the periphery are processed within the central nervous system (CNS) and activate the reflexive effector responses or efferent impulses. These effector responses are designed to expand plasma volume, maintain peripheral perfusion and tissue O2 delivery, and restore homeostasis. The afferent impulses that initiate the body’s intrinsic adaptive responses and converge in the CNS originate from a variety of sources. The initial inciting event usually is loss of circulating blood volume. Other stimuli that can produce the neuroendocrine response include pain, hypoxemia, hypercarbia, acidosis, infection, change in temperature, emotional arousal, or hypoglycemia. The sensation of pain from injured tissue is transmitted via the spinothalamic tracts, resulting in activation of the hypothalamic-pituitary-adrenal axis, as well as activation of the autonomic nervous system (ANS) to induce direct sympathetic stimulation of the adrenal medulla to release catecholamines.
Baroreceptors also are an important afferent pathway in initiation of adaptive responses to shock. Volume receptors, sensitive to changes in both chamber pressure and wall stretch, are present within the atria of the heart. They become activated with low volume hemorrhage or mild reductions in right atrial pressure. Receptors in the aortic arch and carotid bodies respond to alterations in pressure or stretch of the arterial wall, responding to larger reductions in intravascular volume or pressure. These receptors normally inhibit induction of the ANS. When activated, these baroreceptors diminish their output, thus disinhibiting the effect of the ANS. The ANS then increases its output, principally via sympathetic activation at the vasomotor centers of the brain stem, producing centrally mediated constriction of peripheral vessels.
Chemoreceptors in the aorta and carotid bodies are sensitive to changes in O2 tension, H+ ion concentration, and carbon dioxide (CO2) levels. Stimulation of the chemoreceptors results in vasodilation of the coronary arteries, slowing of the heart rate, and vasoconstriction of the splanchnic and skeletal circulation. In addition, a variety of protein and nonprotein mediators are produced at the site of injury as part of the inflammatory response, and they act as afferent impulses to induce a host response. These mediators include histamine, cytokines, eicosanoids, and endothelins, among others that are discussed in greater detail later in this chapter in the Immune and Inflammatory Responses section.
Changes in cardiovascular function are a result of the neuroendocrine response and ANS response to shock, and constitute a prominent feature of both the body’s adaptive response mechanism and the clinical signs and symptoms of the patient in shock. Hemorrhage results in diminished venous return to the heart and decreased cardiac output. This is compensated by increased cardiac heart rate and contractility, as well as venous and arterial vasoconstriction. Stimulation of sympathetic fibers innervating the heart leads to activation of β1-adrenergic receptors that increase heart rate and contractility in this attempt to increase cardiac output. Increased myocardial O2 consumption occurs as a result of the increased workload; thus, myocardial O2 supply must be maintained or myocardial dysfunction will develop. The cardiovascular response in hemorrhage/hypovolemia differs from the responses elicited with the other etiologies of shock. These are compared in Table 5-2.
Direct sympathetic stimulation of the peripheral circulation via the activation of α1-adrenergic receptors on arterioles induces vasoconstriction and causes a compensatory increase in systemic vascular resistance and blood pressure. The arterial vasoconstriction is not uniform; marked redistribution of blood flow results. Selective perfusion to tissues occurs due to regional variations in arteriolar resistance, with blood shunted away from less essential organ beds such as the intestine, kidney, and skin. In contrast, the brain and heart have autoregulatory mechanisms that attempt to preserve their blood flow despite a global decrease in cardiac output. Direct sympathetic stimulation also induces constriction of venous vessels, decreasing the capacitance of the circulatory system and accelerating blood return to the central circulation.
Increased sympathetic output induces catecholamine release from the adrenal medulla. Catecholamine levels peak within 24 to 48 hours of injury and then return to baseline. Persistent elevation of catecholamine levels beyond this time suggests ongoing noxious afferent stimuli. The majority of the circulating epinephrine is produced by the adrenal medulla, while norepinephrine is derived from synapses of the sympathetic nervous system. Catecholamine effects on peripheral tissues include stimulation of hepatic glycogenolysis and gluconeogenesis to increase circulating glucose availability to peripheral tissues, an increase in skeletal muscle glycogenolysis, suppression of insulin release, and increased glucagon release.
The stress response includes activation of the ANS as discussed earlier in the Afferent Signals section, as well as activation of the hypothalamic-pituitary-adrenal axis. Shock stimulates the hypothalamus to release corticotropin-releasing hormone, which results in the release of adrenocorticotropic hormone (ACTH) by the pituitary. ACTH subsequently stimulates the adrenal cortex to release cortisol. Cortisol acts synergistically with epinephrine and glucagon to induce a catabolic state. Cortisol stimulates gluconeogenesis and insulin resistance, resulting in hyperglycemia as well as muscle cell protein breakdown and lipolysis to provide substrates for hepatic gluconeogenesis. Cortisol causes retention of sodium and water by the nephrons of the kidney. In the setting of severe hypovolemia, ACTH secretion occurs independently of cortisol negative feedback inhibition.
The renin-angiotensin system is activated in shock. Decreased renal artery perfusion, β-adrenergic stimulation, and increased renal tubular sodium concentration cause the release of renin from the juxtaglomerular cells. Renin catalyzes the conversion of angiotensinogen (produced by the liver) to angiotensin I, which is then converted to angiotensin II by angiotensin-converting enzyme (ACE) produced in the lung. While angiotensin I has no significant functional activity, angiotensin II is a potent vasoconstrictor of both splanchnic and peripheral vascular beds, and also stimulates the secretion of aldosterone, ACTH, and antidiuretic hormone (ADH). Aldosterone, a mineralocorticoid, acts on the nephron to promote reabsorption of sodium and, as a consequence, water. Potassium and hydrogen ions are lost in the urine in exchange for sodium.
The pituitary also releases vasopressin or ADH in response to hypovolemia, changes in circulating blood volume sensed by baroreceptors and left atrial stretch receptors, and increased plasma osmolality detected by hypothalamic osmoreceptors. Epinephrine, angiotensin II, pain, and hyperglycemia increase production of ADH. ADH levels remain elevated for about 1 week after the initial insult, depending on the severity and persistence of the hemodynamic abnormalities. ADH acts on the distal tubule and collecting duct of the nephron to increase water permeability, decrease water and sodium losses, and preserve intravascular volume. Also known as arginine vasopressin, ADH acts as a potent mesenteric vasoconstrictor, shunting circulating blood away from the splanchnic organs during hypovolemia.11 This may contribute to intestinal ischemia and predispose to intestinal mucosal barrier dysfunction in shock states. Vasopressin also increases hepatic gluconeogenesis and increases hepatic glycolysis.
In septic states, endotoxin directly stimulates arginine vasopressin secretion independently of blood pressure, osmotic, or intravascular volume changes. Proinflammatory cytokines also contribute to arginine vasopressin release. Interestingly, patients on chronic therapy with ACE inhibitors are more at risk of developing hypotension and vasodilatory shock with open heart surgery. Low plasma levels of arginine vasopressin were confirmed in these patients.12
At rest, the majority of the blood volume is within the venous system. Venous return to the heart generates ventricular end-diastolic wall tension, a major determinant of cardiac output. Gravitational shifts in blood volume distribution are quickly corrected by alterations in venous capacity. With decreased arteriolar inflow, there is active contraction of the venous smooth muscle and passive elastic recoil in the thin-walled systemic veins. This increases venous return to the heart, thus maintaining ventricular filling.
Most alterations in cardiac output in the normal heart are related to changes in preload. Increases in sympathetic tone have a minor effect on skeletal muscle beds but produce a dramatic reduction in splanchnic blood volume, which normally holds 20% of the blood volume.
The normal circulating blood volume is maintained within narrow limits by the kidney’s ability to manage salt and water balance with external losses via systemic and local hemodynamic changes and hormonal effects of renin, angiotensin, and ADH. These relatively slow responses maintain preload by altering circulating blood volume. Acute responses to intravascular volume include changes in venous tone, systemic vascular resistance, and intrathoracic pressure, with the slower hormonal changes less important in the early response to volume loss. Furthermore, the net effect of preload on cardiac output is influenced by cardiac determinants of ventricular function, which include coordinated atrial activity and tachycardia.
The Frank-Starling curve describes the force of ventricular contraction as a function of its preload. This relationship is based on force of contraction being determined by initial muscle length. Intrinsic cardiac disease will shift the Frank-Starling curve and alter mechanical performance of the heart. In addition, cardiac dysfunction has been demonstrated experimentally in burns and in hemorrhagic, traumatic, and septic shock.
Afterload is the force that resists myocardial work during contraction. Arterial pressure is the major component of afterload influencing the ejection fraction. This vascular resistance is determined by precapillary smooth muscle sphincters. Blood viscosity also will increase vascular resistance. As afterload increases in the normal heart, stroke volume can be maintained by increases in preload. In shock, with decreased circulating volume and therefore diminished preload, this compensatory mechanism to sustain cardiac output is impeded. The stress response with acute release of catecholamines and sympathetic nerve activity in the heart increases contractility and heart rate.
The microvascular circulation plays an integral role in regulating cellular perfusion and is significantly influenced in response to shock. The microvascular bed is innervated by the sympathetic nervous system and has a profound effect on the larger arterioles. Following hemorrhage, larger arterioles vasoconstrict; however, in the setting of sepsis or neurogenic shock, these vessels vasodilate. Additionally, a host of other vasoactive proteins, including vasopressin, angiotensin II, and endothelin-1, also lead to vasoconstriction to limit organ perfusion to organs such as skin, skeletal muscle, kidneys, and the gastrointestinal (GI) tract to preserve perfusion of the myocardium and CNS.
Flow in the capillary bed is heterogeneous in shock states, which likely is secondary to multiple local mechanisms, including endothelial cell swelling, dysfunction, and activation marked by the recruitment of leukocytes and platelets.13 Together, these mechanisms lead to diminished capillary perfusion that may persist after resuscitation. In hemorrhagic shock, correction of hemodynamic parameters and restoration of O2 delivery generally lead to restoration of tissue O2 consumption and tissue O2 levels. In contrast, regional tissue dysoxia often persists in sepsis, despite similar restoration of hemodynamics and O2 delivery. Whether this defect in O2 extraction in sepsis is the result of heterogeneous impairment of the microcirculation (intraparenchymal shunting) or impaired tissue parenchymal cell oxidative phosphorylation and O2 consumption by the mitochondria is not resolved.14 Interesting data suggest that in sepsis the response to limit O2 consumption by the tissue parenchymal cells is an adaptive response to the inflammatory signaling and decreased perfusion.15
An additional pathophysiologic response of the microcirculation to shock is failure of the integrity of the endothelium of the microcirculation and development of capillary leak, intracellular swelling, and the development of an extracellular fluid deficit. Seminal work by Shires helped to define this phenomenon.8,16 There is decreased capillary hydrostatic pressure secondary to changes in blood flow and increased cellular uptake of fluid. The result is a loss of extracellular fluid volume. The cause of intracellular swelling is multifactorial, but dysfunction of energy-dependent mechanisms, such as active transport by the sodium-potassium pump, contributes to loss of membrane integrity.
Capillary dysfunction also occurs secondary to activation of endothelial cells by circulating inflammatory mediators generated in septic or traumatic shock. This exacerbates endothelial cell swelling and capillary leak, as well as increases leukocyte adherence. This results in capillary occlusion, which may persist after resuscitation, and is termed no-reflow. Further ischemic injury ensues as well as release of inflammatory cytokines to compound tissue injury. Experimental models have shown that neutrophil depletion in animals subjected to hemorrhagic shock produces fewer capillaries with no-reflow and lower mortality.13
METABOLIC EFFECTS
Cellular metabolism is based primarily on the hydrolysis of adenosine triphosphate (ATP). The splitting of the phosphoanhydride bond of the terminal or γ-phosphate from ATP is the source of energy for most processes within the cell under normal conditions. The majority of ATP is generated in our bodies through aerobic metabolism in the process of oxidative phosphorylation in the mitochondria. This process is dependent on the availability of O2 as a final electron acceptor in the electron transport chain. As O2 tension within a cell decreases, there is a decrease in oxidative phosphorylation, and the generation of ATP slows. When O2 delivery is so severely impaired such that oxidative phosphorylation cannot be sustained, the state is termed dysoxia.17 When oxidative phosphorylation is insufficient, the cells shift to anaerobic metabolism and glycolysis to generate ATP. This occurs via the breakdown of cellular glycogen stores to pyruvate. Although glycolysis is a rapid process, it is not efficient, allowing for the production of only 2 mol of ATP from 1 mol of glucose. This is compared to complete oxidation of 1 mol of glucose that produces 38 mol of ATP. Additionally, under hypoxic conditions in anaerobic metabolism, pyruvate is converted into lactate, leading to an intracellular metabolic acidosis.
There are numerous consequences secondary to these metabolic changes. The depletion of ATP potentially influences all ATP-dependent cellular processes. This includes maintenance of cellular membrane potential, synthesis of enzymes and proteins, cell signaling, and DNA repair mechanisms. Decreased intracellular pH also influences vital cellular functions such as normal enzyme activity, cell membrane ion exchange, and cellular metabolic signaling.18 These changes also will lead to changes in gene expression within the cell. Furthermore, acidosis leads to changes in calcium metabolism and calcium signaling. Compounded, these changes may lead to irreversible cell injury and death.
Epinephrine and norepinephrine have a profound impact on cellular metabolism. Hepatic glycogenolysis, gluconeogenesis, ketogenesis, skeletal muscle protein breakdown, and adipose tissue lipolysis are increased by catecholamines. Cortisol, glucagon, and ADH also contribute to the catabolism during shock. Epinephrine induces further release of glucagon, while inhibiting the pancreatic β-cell release of insulin. The result is a catabolic state with glucose mobilization, hyperglycemia, protein breakdown, negative nitrogen balance, lipolysis, and insulin resistance during shock and injury. The relative underuse of glucose by peripheral tissues preserves it for the glucose-dependent organs such as the heart and brain.
Hypoperfused cells and tissues experience what has been termed oxygen debt, a concept first proposed by Crowell in 1961.19 The O2 debt is the deficit in tissue oxygenation over time that occurs during shock. When O2 delivery is limited, O2 consumption can be inadequate to match the metabolic needs of cellular respiration, creating a deficit in O2 requirements at the cellular level. The measurement of O2 deficit uses calculation of the difference between the estimated O2 demand and the actual value obtained for O2 consumption. Under normal circumstances, cells can “repay” the O2 debt during reperfusion. The magnitude of the O2 debt correlates with the severity and duration of hypoperfusion. Surrogate values for measuring O2 debt include base deficit and lactate levels and are discussed later in the Hypovolemic/Hemorrhagic section.
In addition to induction of changes in cellular metabolic pathways, shock also induces changes in cellular gene expression. The DNA binding activity of a number of nuclear transcription factors is altered by hypoxia and the production of O2 radicals or nitrogen radicals that are produced at the cellular level by shock. Expression of other gene products such as heat shock proteins, vascular endothelial growth factor, inducible nitric oxide synthase (iNOS), heme oxygenase-1, and cytokines also are clearly increased by shock.20 Many of these shock-induced gene products, such as cytokines, have the ability to subsequently alter gene expression in specific target cells and tissues. The involvement of multiple pathways emphasizes the complex, integrated, and overlapping nature of the response to shock.
IMMUNE AND INFLAMMATORY RESPONSES
The inflammatory and immune responses are a complex set of interactions between circulating soluble factors and cells that can arise in response to trauma, infection, ischemia, toxic, or autoimmune stimuli.20 The processes are well regulated and can be conceptualized as an ongoing surveillance and response system that undergoes a coordinated escalation following injury to heal disrupted tissue or restore host-microbe equilibrium, as well as active suppression back to baseline levels. Failure to adequately control the activation, escalation, or suppression of the inflammatory response can lead to systemic inflammatory response syndrome and potentiate multiple organ failure.
Both the innate and adaptive branches of the immune system work in concert to rapidly respond in a specific and effective manner to challenges that threaten an organism’s well-being. Each arm of the immune system has its own set of functions, defined primarily by distinct classes of effector cells and their unique cell membrane receptor families. Alterations in the activity of the innate host immune system can be responsible for both the development of shock (i.e., septic shock following severe infection and traumatic shock following tissue injury with hemorrhage) and the pathophysiologic sequelae of shock such as the proinflammatory changes seen following hypoperfusion (see Fig. 5-1). When the predominantly paracrine mediators gain access to the systemic circulation, they can induce a variety of metabolic changes that are collectively referred to as the host inflammatory response. Understanding of the intricate, redundant, and interrelated pathways that comprise the inflammatory response to shock continues to expand. Despite limited understanding of how our current therapeutic interventions impact the host response to illness, inappropriate or excessive inflammation appears to be an essential event in the development of ARDS, multiple organ dysfunction syndrome (MODS), and posttraumatic immunosuppression that can prolong recovery.21
Following direct tissue injury or infection, there are several mechanisms that lead to the activation of the active inflammatory and immune responses. These include release of bioactive peptides by neurons in response to pain and the release of intracellular molecules by broken cells, such as heat shock proteins, mitochondrial products, heparan sulfate, high mobility group box 1, and RNA. Only recently has it been realized that the release of intracellular products from damaged and injured cells can have paracrine and endocrine-like effects on distant tissues to activate the inflammatory and immune responses.22 This hypothesis, which was first proposed by Matzinger, is known as danger signaling. Under this novel paradigm of immune function, endogenous molecules are capable of signaling the presence of danger to surrounding cells and tissues. These molecules that are released from cells are known as damage-associated molecular patterns (DAMPs) (Table 5-3). DAMPs are recognized by cell surface receptors to effect intracellular signaling that primes and amplifies the immune response. These receptors are known as pattern recognition receptors (PRRs) and include the Toll-like receptors (TLRs) and the receptor for advanced glycation end products. Interestingly, TLRs and PRRs were first recognized for their role in signaling as part of the immune response to the entry of microbes and their secreted products into a normally sterile environment. These bacterial products, including lipopolysaccharide, are known as pathogen-associated molecular patterns. The salutary consequences of PRR activation most likely relate to the initiation of the repair process and the mobilization of antimicrobial defenses at the site of tissue disruption. However, in the setting of excessive tissue damage, the inflammation itself may lead to further tissue damage, amplifying the response both at the local and systemic level.20 PRR activation leads to intracellular signaling and release of cellular products including cytokines (Fig. 5-4).
Mitochondrial DNA Hyaluronan oligomers Heparan sulfate Extra domain A of fibronectin Heat shock proteins 60, 70, Gp96 Surfactant Protein A β-Defensin 2 Fibrinogen Biglycan High mobility group box 1 Uric acid Interleukin-1α S-100s Nucleolin |
Figure 5-4.
A schema of information flow between immune cells in early inflammation following tissue injury and infection. Cells require multiple inputs and stimuli before activation of a full response. DAMPs = damage-associated molecular patterns; HMGB1 = high mobility group box 1; TNF = tumor necrosis factor.
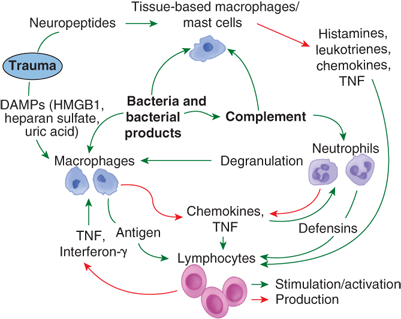
Before the recruitment of leukocytes into sites of injury, tissue-based macrophages or mast cells act as sentinel responders, releasing histamines, eicosanoids, tryptases, and cytokines (Fig. 5-5). Together these signals amplify the immune response by further activation of neurons and mast cells, as well as increasing the expression of adhesion molecules on the endothelium. Furthermore, these mediators cause leukocytes to release platelet-activating factor, further increasing the stickiness of the endothelium. Additionally, the coagulation and kinin cascades impact the interaction of endothelium and leukocytes.
Figure 5-5.
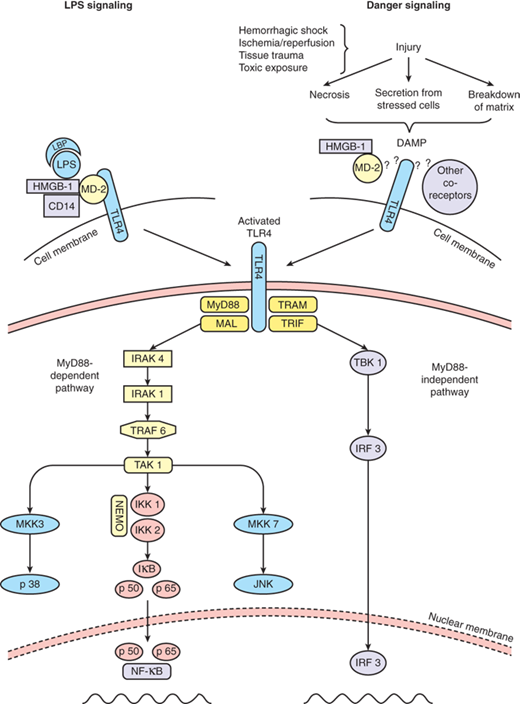
Signaling via the pattern recognition receptor TLR4. LPS signaling via TLR4 requires the cofactors LPS binding protein (LBP), MD-2, and CD14. Endogenous danger signals released from a variety of sources also signal in a TLR4-dependent fashion, although it is as yet unknown what cofactors may be required for this activity. Once TLR4 is activated, an intracellular signaling cascade is initiated that involves both a MyD88-dependent and independent pathway. DAMP = damage-associated molecular pattern; LPS = lipopolysaccharide; MD-2 = myeloid differentiation factor-2; MyD88 = myeloid differentiation primary response gene 88; NF-κB = nuclear factor-κB; TLR4 = Toll-like receptor-4. (Reproduced with permission from Mollen KP, Anand RJ, Tsung A, et al.83 Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock. 2006;26:430–437.)
The immune response to shock encompasses the elaboration of mediators with both proinflammatory and anti-inflammatory properties (Table 5-4). Furthermore, new mediators, new relationships between mediators, and new functions of known mediators are continually being identified. As new pathways are uncovered, understanding of the immune response to injury and the potential for therapeutic intervention by manipulating the immune response following shock will expand. What seems clear at present, however, is that the innate immune response can help restore homeostasis, or if it is excessive, promote cellular and organ dysfunction.
Multiple mediators have been implicated in the host immune response to shock. It is likely that some of the most important mediators have yet to be discovered, and the roles of many known mediators have not been defined. A comprehensive description of all of the mediators and their complex interactions is beyond the scope of this chapter. For a general overview, a brief description of the more extensively studied mediators, and some of the known effects of these substances, see the discussion below. A more comprehensive review can be found in Chap. 2.
Tumor necrosis factor alpha (TNF-α) was one of the first cytokines to be described and is one of the earliest cytokines released in response to injurious stimuli. Monocytes, macrophages, and T cells release this potent proinflammatory cytokine. TNF-α levels peak within 90 minutes of stimulation and return frequently to baseline levels within 4 hours. Release of TNF-α may be induced by bacteria or endotoxin and leads to the development of shock and hypoperfusion, most commonly observed in septic shock. Production of TNF-α also may be induced following other insults, such as hemorrhage and ischemia. TNF-α levels correlate with mortality in animal models of hemorrhage.23 In contrast, the increase in serum TNF-α levels reported in trauma patients is far less than that seen in septic patients.24 Once released, TNF-α can produce peripheral vasodilation, activate the release of other cytokines, induce procoagulant activity, and stimulate a wide array of cellular metabolic changes. During the stress response, TNF-α contributes to the muscle protein breakdown and cachexia.
Interleukin-1 (IL-1) has actions similar to those of TNF-α. IL-1 has a very short half-life (6 min) and primarily acts in a paracrine fashion to modulate local cellular responses. Systemically, IL-1 produces a febrile response to injury by activating prostaglandins in the posterior hypothalamus, and causes anorexia by activating the satiety center. This cytokine also augments the secretion of ACTH, glucocorticoids, and β-endorphins. In conjunction with TNF-α, IL-1 can stimulate the release of other cytokines such as IL-2, IL-4, IL-6, IL-8, granulocyte-macrophage colony-stimulating factor, and interferon-γ.
IL-2 is produced by activated T cells in response to a variety of stimuli and activates other lymphocyte subpopulations and natural killer cells. The lack of clarity regarding the role of IL-2 in the response to shock is intimately associated with that of understanding immune function after injury. Some investigators have postulated that increased IL-2 secretion promotes shock-induced tissue injury and the development of shock. Others have demonstrated that depressed IL-2 production is associated with, and perhaps contributes to, the depression in immune function after hemorrhage that may increase the susceptibility of patients who develop shock to suffer infections.25,26 It has been postulated that overly exuberant proinflammatory activation promotes tissue injury, organ dysfunction, and the subsequent immune dysfunction/suppression that may be evident later.21 Emphasizing the importance of temporal changes in the production of mediators, both the initial excessive production of IL-2 and later depressed IL-2 production are probably important in the progression of shock.
IL-6 is elevated in response to hemorrhagic shock, major operative procedures, or trauma. Elevated IL-6 levels correlate with mortality in shock states. IL-6 contributes to lung, liver, and gut injury after hemorrhagic shock.27 Thus, IL-6 may play a role in the development of diffuse alveolar damage and ARDS. IL-6 and IL-1 are mediators of the hepatic acute phase response to injury; enhance the expression and activity of complement, C-reactive protein, fibrinogen, haptoglobin, amyloid A, and α1-antitrypsin; and promote neutrophil activation.28
IL-10 is considered an anti-inflammatory cytokine that may have immunosuppressive properties. Its production is increased after shock and trauma, and it has been associated with depressed immune function clinically, as well as an increased susceptibility to infection.29 IL-10 is secreted by T cells, monocytes, and macrophages, and inhibits proinflammatory cytokine secretion, O2 radical production by phagocytes, adhesion molecule expression, and lymphocyte activation.29,30 Administration of IL-10 depresses cytokine production and improves some aspects of immune function in experimental models of shock and sepsis.31,32
Recent studies point to the importance of chemokines, a specific set of cytokines, that have the ability to induce chemotaxis of leukocytes. Chemokines bind to specific chemokine receptors and transduce chemotactic signals to leukocytes. The significance of this large family of chemoattractant cytokines in immunology is difficult to understate, as almost every facet of the immune system is influenced by chemokines, including immune system development, immune surveillance, immune priming, effector responses, and immune regulation.33
The complement cascade can be activated by injury, shock, and severe infection, and contributes to host defense and proinflammatory activation. Significant complement consumption occurs after hemorrhagic shock.34 In trauma patients, the degree of complement activation is proportional to the magnitude of injury and may serve as a marker for severity of injury. Patients in septic shock also demonstrate activation of the complement pathway, with elevations of the activated complement proteins C3a and C5a. Activation of the complement cascade can contribute to the development of organ dysfunction. Activated complement factors C3a, C4a, and C5a are potent mediators of increased vascular permeability, smooth muscle cell contraction, histamine and arachidonic acid by-product release, and adherence of neutrophils to vascular endothelium. Activated complement acts synergistically with endotoxin to induce the release of TNF-α and IL-1. The development of ARDS and MODS in trauma patients correlates with the intensity of complement activation.35 Complement and neutrophil activation may correlate with mortality in multiply injured patients.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


