Do not put your faith in what statistics say until you have carefully considered what they do not say.
–William W. Watt
When something can be expressed in a numerical way, it is an aid to more precise and accurate thinking.
–Richard Ascher
Absence of evidence is not evidence of absence.
–Anonymous
Because statisticians view numbers (i.e., data) from many perspectives and usually can evaluate data with different approaches, depending on the factors and contexts involved, it has given rise to various jokes. One old joke concerns three people who were asked the sum of two plus two. The first was a banker and he answered “four.” An accountant was second, and he gave the same response. But, when a statistician was asked the sum of two plus two, he took the questioner aside before responding in a whisper: “What do you want it to equal?” The point of mentioning this anecdote is not to suggest that statisticians manipulate data, but that there are usually numerous ways in which to evaluate a set of data.
It should not be necessary to discuss or even to say the obvious, that all professionals in drug discovery, development,
marketing, production, and in many other functions as well must work closely with knowledgeable and competent statisticians to discover and develop new therapies, diagnostics, and other healthcare products. This chapter, however, focuses on clinical issues and provides a clinical perspective on a number of statistical issues.
At the outset of this chapter, the author must state categorically that he does not have the expertise to write a chapter about statistics. Therefore, this is not a chapter about statistics, but rather a collection of some statistical issues that are of particular interest to this author from a clinical perspective. Other fascinating statistical issues, such as Simpson’s Paradox and regression to the mean are discussed in
Guide to Clinical Trials (
Spilker 1991) and will not be repeated here.
ROLES OF STATISTICS AND STATISTICIANS IN DRUG DEVELOPMENT
It is useful to broadly describe the universe of statistics in drug discovery and development. In the preclinical discovery area, scientists utilize statistical principles and methods in the design, execution, analysis, and interpretation of their experiments. The statistics department or a consulting statistician should be an integral part of all preclinical scientific teams conducting experiments, not only those where sophisticated analyses or concepts are required. Statisticians may also act as consultants to departments that need advice or assistance with experimental design, data analyses, data automation, or other functions.
The largest role of statistics in the pharmaceutical industry in terms of assignment of personnel is in the clinical area. Regardless of whether clinical statisticians are organizationally part of the clinical trials group or report to another part of the organization, there must be close interactions between statisticians and clinical trial personnel. In the areas of clinical trials, these interactions focus primarily on trial design, protocol development, data analyses, and final medical report writing. In addition, statisticians assist in determining the appropriate data to collect and also help devise an appropriate “trial-specific” data management process for trials where that is necessary. Statisticians are also integral parts of virtually all project teams developing drugs and all Data Safety Monitoring Boards (
DSMBs), and are on many scientific advisory boards.
Statisticians are often asked to analyze special problems or issues that arise during clinical trials, and are usually asked to attend regulatory meetings. In some companies, statisticians are a resource to others and receive their work assignments from them, rather than being independent contributors to the teams’ objectives. Statisticians are usually based in a single research and development department, but may be placed organizationally in multiple departments and functions, including preclinical discovery, clinical, and marketing, so that they are closer to the people they are interacting with.
Another major area where statistics has intimate involvement is in the quality assurance area of production. A number of the roles played by statisticians in quality assurance are described in
Chapter 108, but, of course, other activities of statisticians (in all other functional areas) are essential to the well being of the company.
STATISTICAL ISSUES INVOLVING CLINICAL TRIALS: PRETRIAL
Trial Design
Only a few general comments are offered on this large topic. The main principles are as follows:
Statisticians and clinicians must both be involved in creating the most appropriate clinical trial designs.
They must have mutual respect and a positive working relationship for the trial design to be smoothly prepared and reviewed.
When clinicians are not familiar with the basics of statistics an effort should be made to introduce them to those principles and methods.
It is also important for the statistician to have at least basic knowledge of the therapeutic area, disease, and drug profile.
The timing when a statistician is to be involved in basic design considerations, generating the statistical portion of a protocol, creating the statistical analysis plan (
SAP), and other activities must be understood by the clinicians authoring and managing the clinical trial.
Statisticians should be asked to review and provide input to the clinical trial protocol at several points during its generation.
Is It Required for Statisticians to Provide Input and Review All Clinical Protocols?
Over the past 40 years, the pendulum has swung from an almost total “no” to a virtually complete “yes,” primarily because this represents good science and is cited in the International Conference on Harmonisation guidelines. There are few pharmaceutical companies, even those outside the developed countries, where this practice is not required.
While the work that statisticians provide in creating the sample size estimates for a clinical trial are among their most well-known activities, their main contribution is in overall study design and analysis strategies.
Sample Size Estimation
A basic principle is that a clinical trial will require larger sample sizes to detect small expected differences between treatment groups and smaller sample sizes to detect larger differences. Sample sizes are usually determined by statisticians on the basis of:
The magnitude of the effect that is important for the trial to detect
The variability (often estimated) of the primary variable
The desired probability (power) of observing that effect (or a more “extreme” effect) with a defined significance level
The acceptable type I error (i.e., level of significance) (usually 0.05)
A power of 80% was often chosen as adequate for most controlled trials in the 1980s and 1990s, but the trend has been to move to 90%. Some companies prefer to use even higher numbers (e.g., 95%) to ensure a greater probability of detecting an effect if one is present and to have a greater chance of using a single large study as a “one-study submission.”
While power is maximized by having the same numbers of patients in each treatment group, this may be modified when it would affect enrollment, or if a company wished to obtain more safety data on the test drug. For instance, in testing analgesics almost all patients will want to know that they have greater than a 50% chance of receiving the active therapy or an active control (versus the possibility of receiving a placebo) before they will be willing to enroll in a trial. Therefore, in a clinical trial of an analgesic it is highly desirable to offer patients more than a 50-50 chance of receiving the active treatment being tested, and unequal size groups are created. Unless this is done it is highly probable that recruitment will be negatively affected and in chronic studies that retention will also be negatively affected. Usually, unequal size groups of two to one or even three to one do not result in a major loss of power and may be acceptable, but any higher ratios between the groups usually will not be acceptable due to a loss of power.
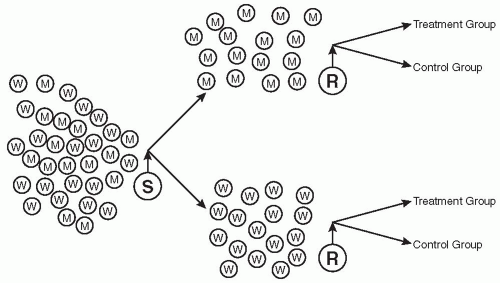
Stratification of Patients
Whenever there are known (or believed) to be differences in how certain groups of people or certain factors will influence the outcome of a trial, i.e., how two or more types of patients will respond to treatment, it is important to group each type separately to reduce variability and to be better able to discern an effect of the treatment if one is present. Mixing two or more groups who are likely to respond differently to therapy (e.g., those with mild disease and those with severe disease) is likely to decrease the overall response to a treatment and may prevent a positive response from being observed. The most widely used approaches are to either conduct a separate trial in each group of patients or to stratify them prior to randomization. This is illustrated in
Fig. 101.1. However, one may also evaluate differential effects of groups of patients as part of the analysis if this analysis is pre-planned in the protocol’s
SAP.
Some of the more commonly encountered groups that may respond differently to treatments are men versus women, children versus young adults versus geriatric adults, patients with mild disease versus those with moderate or severe disease, one race versus another, those with different risk factors or different numbers of risk factors (e.g., for a myocardial infarction), or those with one etiology of their disease versus those with another (e.g., those with back pain from muscular causes versus those whose cause is neurological). Stratification reduces variability, which increases power and also permits evaluation of the stratification factor and its influence on the outcome. This allows one to assess whether or not differences between the groups stratified exist and if that difference affects their responses to treatment. It is assumed that one has considered the number of patients to include in each group and that there are sufficient numbers enrolled and treated.
When stratification is not done a priori, then one may wish to analyze data separately for the two groups that may have responded differently to treatment. However, such subgroup analyses conducted post hoc are not generally considered as valid by regulatory agencies as a basis for a drug’s registration, but is an important basis for forming a hypothesis that may be tested in another trial.
Another reason to stratify patients is that by chance alone an excess number of one type or group of patients may be randomized to receive placebo (or another control) and this may cause a substantial problem in skewing the data obtained and possibly making the entire trial uninterpretable. One case occurred when a beta blocker was being compared with placebo for a possible influence on the rate of myocardial re-infarction when the drug was given for a full year. It turned out by chance that the risk factors
for re-infarction were quite different between the two groups of patients and this was not used as a stratification method, nor was the number or extent of risk factors used as an inclusion criterion for the trial. The data were deemed uninterpretable and the trial and the enormous efforts expended were wasted.
Enrolling More Patients in a Clinical Trial Than Approved by an Institutional Review Board/Ethics Committee
A protocol may include an interim analysis to evaluate the question of whether it is important to enroll more patients than planned. For example, if a trial is based on a treatment decreasing the number of occurrences of the primary endpoint in a population, (e.g., number of seizures, number of migraine attacks, number of myocardial infarctions) but the number observed in the trial turn out to be fewer than anticipated, additional patients will be required to evaluate if an effect is present. In this situation, there are several alternatives.
One alternative is that one can hope that the number of events will increase in the remainder of the trial. A second alternative would be to approach the regulatory agency and tell them that the assumptions of the original sample size were in error and the company is compelled to increase the number of patients or to alter the trial’s design. In that situation, the company may propose an interim analysis (that was not originally planned) to assess how the “n” of the trial has to be modified.
A third alternative might be that the possibility of this lowered event rate may have been anticipated in advance and carefully defined rules established prior to initiating the trial created to re-evaluate the number of patients needed to be enrolled during a scheduled interim analysis. These rules are then used, often by a
DSMB using interim data to make a decision on resizing the trial. In general, if 10% or fewer additional patients are to be enrolled, a protocol amendment is not usually required, but if the number exceeds 10% an Institutional Review Board/Ethics Committee approval will generally be required. An important point for double-blind trials is that this decision and action must be completed prior to breaking the blind.
Statistical Analysis Plans
In the protocol there is often a rather short statistical analysis section that describes the statistical considerations in broad terms, i.e., what the company plans to analyze and how this will be accomplished. However, this is not what is referred to as the
SAP. The
SAP is a more detailed description of the specific statistical approaches to be followed and often is approximately 12 pages when prepared for regulatory agencies. It focuses on the types of data expected (e.g., continuous or discontinuous, full of omissions or almost complete) plus the approaches to analyzing the important variables in the trial. Specific table and listing formats are usually not included in the
SAP submitted to regulatory agencies, but are included in a full
SAP, particularly if a contract research organization (
CRO) is involved. The full
SAP discusses the types of analyses that will be conducted for each type of data (i.e., laboratory data, efficacy endpoints, various safety tests). This detailed
SAP is not submitted to regulatory agencies until shortly prior to the database lock. Of course, if the nature and distribution of the data obtained is not what was expected then other statistical approaches are often used. This should be documented before breaking the blind. The
SAP should state, or at least indicate, what will be done when the methodological assumptions do not hold.
There is another type of
SAP that is prepared by a sponsor,
CRO, or other vendor. This is a far more extensive presentation of the many specific tables, figures, and listings that are expected to be created (often well in excess of a hundred). A great deal of time is often spent ensuring that this plan is correct and as polished as possible. This type of
SAP is usually created long before the trial is completed. An example of a table of contents of this type of
SAP is given in
Table 101.1 and a selection of more specific issues in
Table 101.2. Both types of
SAP must be completed before the study is unblinded. This means that a
SAP for an open-label study must be completed before the first patient is enrolled. Nonetheless, many times, it is allowed for the
SAP to be developed up until the time that the database is locked.
Data Imputation
Data imputation is a simple concept that missing data are filled in by one of several methods. The most commonly used method is to average the data obtained prior to the missing data with the data obtained at the next visit. For example, if a patient misses a laboratory examination of a blood chemistry profile of 15 analytes, then each of the missing data points is filled in by averaging the same analyte’s value for the time before and the time after the missing one and the average is used for the missing value. While there is certain logic to this procedure, it is creating data that do not exist and while it facilitates various statistical approaches, which otherwise might not be able to be used, it still is not comfortable for most people to use this approach. Nonetheless, there are many times when such imputations of data are used or even required (e.g., by a regulatory agency).
The Last Observation Carried Forward approach for filling in data of patients who have left the trial early for any reason is another type of imputation of data and, again, is not one that many people (both statisticians and clinicians) want to do. This approach, however, is often used and is even suggested or required by some regulatory agencies.