Progressive Familial Intrahepatic Cholestasis
Joseph Misdraji, MD
Key Facts
Etiology/Pathogenesis
PFIC1 (FIC1 disease) is due to mutations of ATP8B1 (FIC1 gene)
PFIC2 (BSEP disease) is due to mutations of ABCB11 gene that encodes bile salt export pump
PFIC3 is due to mutation of ABCB4 gene that encodes MDR3, a phospholipid flippase
Clinical Issues
PFIC presents in 1st year of life with intense pruritus and jaundice
PFIC1 and PFIC2 are characterized by normal serum GGT, whereas PFIC3 is associated with elevated GGT
Progressive forms lead to liver failure, cirrhosis, and death before adulthood
Partial external biliary diversion or ileal exclusion may be used with some success, but most patients require liver transplantation
Microscopic Pathology
PFIC1 is characterized by relatively bland canalicular cholestasis
PFIC2 is characterized by pattern of neonatal giant cell hepatitis
PFIC3 is characterized by duct proliferation and bile plugs in ductules
Ancillary Tests
FIC1 disease reveals coarse, granular bile, referred to as Byler bile
Immunohistochemistry for canalicular proteins can be used for diagnosis
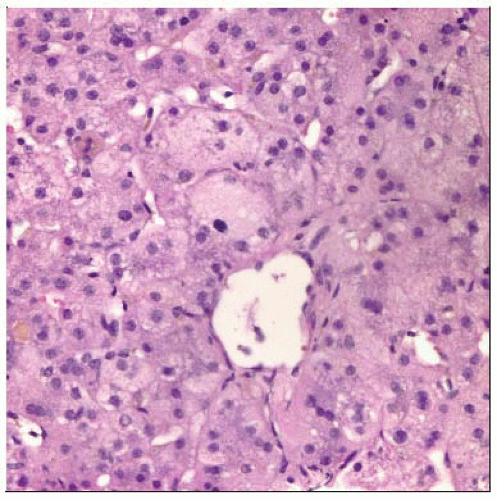 Hematoxylin & eosin stained section of a liver biopsy in a child with PFIC shows giant cell transformation of perivenular hepatocytes, typical of childhood cholestasis syndromes. |
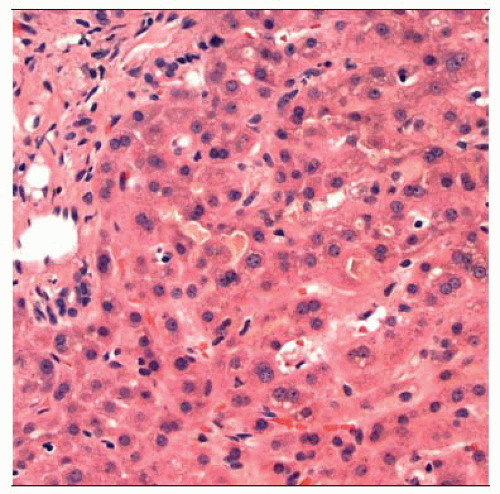 Hematoxylin & eosin stained section of a liver biopsy in an adult patient with BRIC shows bland canalicular cholestasis with mild lobular architectural disarray but minimal inflammation. |
TERMINOLOGY
Abbreviations
Progressive familial intrahepatic cholestasis (PFIC)
Synonyms
PFIC type 1
Familial intrahepatic cholestasis 1 (FIC1) disease
Byler disease, Byler syndrome
Greenland familial cholestasis (GFC)
PFIC type 2
Bile salt export pump (BSEP) disease
PFIC type 3
Multidrug resistance protein 3 (MDR3) disease
Definitions
Heterogeneous group of autosomal recessive disorders characterized by chronic cholestasis and progression to cirrhosis and liver failure
ETIOLOGY/PATHOGENESIS
Autosomal Recessive Genetic Disorder
PFIC1
Mutation of ATP8B1 (FIC1 gene), located on chromosome 18q21-q22
FIC1 is expressed on variety of tissues including liver, intestine, pancreas
Functions as aminophospholipid flippase, flipping phosphatidylserine from outer to inner lipid layer of cell membrane
Mechanism of cholestasis unclear
PFIC2
Mutations of ABCB11 gene on chromosome 2q24 that encodes BSEP, an ATP-dependent bile acid transporter on canalicular membrane
PFIC3
Mutation of ABCB4 gene that encodes MDR3 glycoprotein
MDR3 is flippase that flips phosphatidylcholine from inner to outer lipid leaflet of canalicular membrane
Phosphatidylcholine in bile reduces its detergent action, and MDR3 deficiency results in bile with more detergent properties
Absence of phospholipids destabilizes micelles, promoting lithogenicity of bile with crystallization of cholesterol and leads to small bile duct obstruction
CLINICAL ISSUES
Presentation
FIC1 deficiency disease
Depending on nature of mutation, may present as benign recurrent intrahepatic cholestasis (BRIC1) or progressive and severe form (PFIC1)
PFIC1
Presents in 1st year of life with intense pruritus and jaundice
Systemic disorder with extrahepatic manifestations including pancreatitis, diarrhea, respiratory symptoms, failure to thrive, delayed sexual development, hearing loss
BRIC1
Recurrent episodes of cholestasis with intense pruritus
Episodes resolve spontaneously without histologic progression
BSEP disease
Depending on nature of mutation, may present as BRIC2 or PFIC2
PFIC2
Presents as severe intrahepatic cholestasis in infancy
BRIC2
Presents as recurrent episodes of pruritus, steatorrhea, nausea, vomiting, anorexia, right upper quadrant abdominal pain, and weight loss
Frequently complicated by cholesterol cholelithiasis
MDR3 disease
PFIC3 presents during infancy with pruritus, jaundice, pale stools, hepatomegaly, or complications of portal hypertension, such as splenomegaly or gastrointestinal bleeding
MDR3 mutations also seen in patients with intrahepatic lithiasis, cholesterol gallstone disease, intrahepatic cholestasis of pregnancy, transient neonatal cholestasis, cholestatic drug reactions
Laboratory Tests
GGT
Normal in PFIC1 and PFIC2
Elevated in PFIC3
Elevated serum bile acids in all 3 types
PFIC3 is characterized by low concentrations of phospholipids in bile analysis
Natural History
Progressive forms can result in worsening hepatic function, liver failure, cirrhosis, and death before adulthood
Chronic cholestasis leads to complications of fat malabsorption such as deficiencies of fat-soluble vitamins and weight loss
BSEP disease is associated with development of hepatocellular carcinoma
Treatment
Surgical approaches
Partial external biliary diversion or cholecystojejunocutaneostomy
Short jejunal segment is anastomosed to the dome of gallbladder and terminates as stoma, allowing bile to be discarded
Ileal exclusion
Approximately 15% of terminal ileum is bypassed, which reduces bile acid reabsorption
Liver transplantation
May result in intractable diarrhea and steatohepatitis in FIC1 patients
Drugs
Ursodeoxycholic acid (UDCA), rifampin, cholestyramine, and phenobarbital have been used to treat pruritus
MICROSCOPIC PATHOLOGY
Histologic Features
FIC1 disease
BRIC1
Bland canalicular cholestasis with variable cholestatic rosettes, compact hepatocytes, hepatocyte multinucleation without prominent giant cell transformation
Minimal inflammation
PFIC1: Increasing portal fibrosis with eventual cirrhosis
Pattern of fibrosis that involves early pericentral sclerosis, central-to-portal fibrosis, and cirrhosis with lacy lobular fibrosis has been described
Bile duct injury and paucity may be present as well as bile ductular reaction
BSEP disease
Neonatal hepatitis pattern with giant cell transformation, ballooning, inflammation, and canalicular cholestasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


