New products
Transferring products from another plant or manufacturer
Modifying products under change control
Providing contract manufacturing for other companies
Making engineering or equipment modifications
Adding new technologies
Expansion of existing facilities
Table 108.1 Selected production activities to support various projects | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 108.2 Production and other groups that are often on a production project teama | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
A new facility maybe built to focus primarily or exclusively on the new drug. However, this rarely occurs in practice due to the significant resources needed and the potential impact if the product does not meet expectations. However, an active pharmaceutical ingredient (API; i.e., the active substance that has a positive pharmaceutical effect in patients) sometimes requires its own manufacturing facility, either due to safety or the complexity of the process
An existing facility may be “beefed-up” to handle the drug. This is usually more cost effective than the above alternative.
Several existing company manufacturing facilities may each handle different parts of the manufacturing. APIs are typically made in a chemical (or biological) manufacturing site, whereas the filling and finishing normally are separated from the synthetic production due to the higher level of cleanliness and different types of operations, equipment, and levels of technical knowledge required by the operators.
Part or all of the manufacturing may be contracted out to other companies. This is typical in the current environment because many pharmaceutical companies have stated that manufacturing is no longer a core competency. In addition, start-up companies must almost always depend on contractors to manufacture their drugs because they do not have sufficient resources and expertise required to build and operate a manufacturing plant.
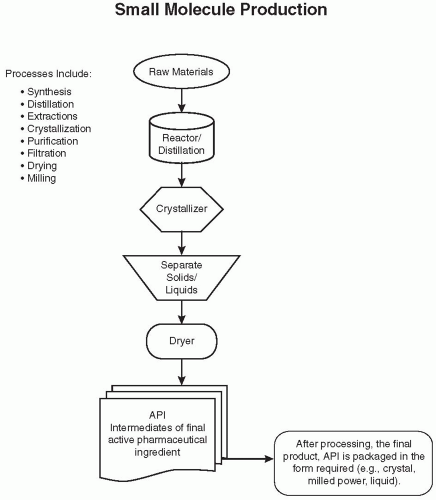 Figure 108.1 Flow diagram of an overview of the chemical production of a hypothetical bulk drug API. These steps are often repeated with additional raw materials or intermediates. (Reprinted from Charlton W, Ingallinera T, Shive D. Validation of clinical manufacturing. In: Agalloco JP, Carleton F, eds. Validation of pharmaceutical processes. 3rd ed. New York: Informa Healthcare; 2007 with permission.) |
waiting for analytical results prior to compressing the granulated product into tablets. Further processing may include adding a sugar coat or film coat over the product. This coating enhances the product’s appearance and taste and, for some, helps with swallowing. It may also be applied to delay absorption, either to create a sustained-release formulation or as an enteric coating to allow the drug to pass through the stomach without dissolving. The product is tested and passed prior to moving to the fill/finish shop floor.
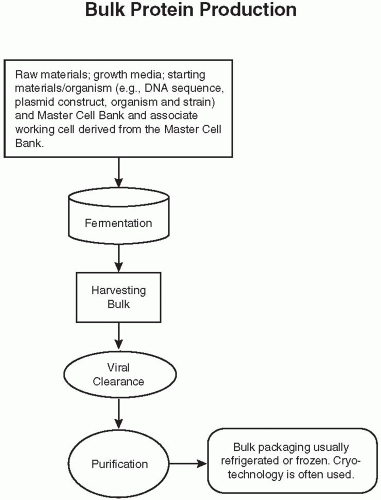 Figure 108.2 Flow diagram of the bulk protein production of a biological. DNA, deoxyribonucleic acid. (Reprinted from Charlton W, Ingallinera T, Shive D. Validation of clinical manufacturing. In: Agalloco JP, Carleton F, eds. Validation of pharmaceutical processes. 3rd ed. New York: Informa Healthcare; 2007 with permission.) |
Bottle descrambler—This is a continuous motion machine that orients the bottle to an upright position and then places the empty bottle on a continuous conveyor. The bottle then proceeds to the next machine.
Bottle filler—This machine fills the product into a bottle. A method to ensure that each bottle has the correct count (tablets) or weight (liquids) is included in this equipment. An alternative for tablets is filling into formed trays or packets that are then sealed. An additional weighing check is often required within the line.
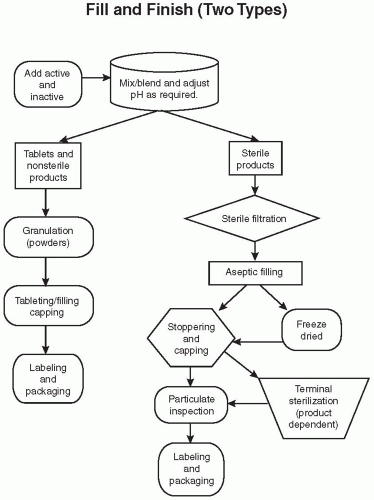
Figure 108.3 Flow diagram of the general fill and finish operations of a sterile and nonsterile product. (Reprinted from Charlton W, Ingallinera T, Shive D. Validation of clinical manufacturing. In: Agalloco JP, Carleton F, eds. Validation of pharmaceutical processes. 3rd ed. New York: Informa Healthcare; 2007 with permission.)
Bottle capper—A bottle capper accepts the bottle from the bottle filler and places caps onto the bottles, usually after cotton and or a desiccant is placed in the bottle.
Cap retorquer—This machine tightens the caps on the bottle one last time.
Bottle labeler—The labeler prints a label and places it on the bottle. The drug product insert is either attached to the bottle or added later into the product carton.
Cartner—The cartner places the bottles and label inserts into cartons.
Case palletizer—The case palletizer collates the boxes into rows, allowing a robotic arm to pick up the rows of boxes and place them on a pallet.
Aseptic filling—The ingredients, components, and process are sterilized, and the product is sterile filtered and filled.
Terminal sterilization—The product in the container closure is sterilized by a steam autoclave or radiation at the end of the production.
Liquids (solutions and suspensions) for eye and ear—Products usually have a closed dispensing method to minimize the exposure of the product to air and sometimes light. Some products are filled in a plastic form and are then sealed, while others are placed in glass vials with droppers or plastic drop dose bottles.
Liquid injectables—The product may be filled into ampoules, vials, form-fill seal, or syringes.
Powders—Filled into vials or depots. Depots are deposited into the body, often just under the skin for administration of a drug for a period of weeks or months.
Freeze-dried products—These are liquid products that are filled into a vial and then lyophilized (i.e., the solvent, usually water, is removed during this process to form a dried cake that enhances the products stability). This process is sometime used with dual-chamber syringes. The first chamber would have the freeze-dried cake, and the second would contain the solvent (e.g., water). When the syringe is activated, the water and cake will mix prior to being injected.
Ophthalmic ointments
Inhalants
Table 108.3 Different standards for number of allowable particles in air in clean and sterile environments | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Sterile gowning—Rooms are designed to handle the environmental load of people entering and gowning with methods that assure the least contamination possible. All employees who are approved to enter the Grade A areas must go through training and testing that verify their capability to properly gown. Periodically, without notice and even after being approved, employees are tested as they enter, as they perform clean room responsibilities, and also as they leave to check whether they could have impacted the batch.
Water for injection—Water defined in the US Pharmacopeia (USP) and manufactured normally by distillation, but other methods are also available such as reverse osmosis and ultra filtration.
Pure steam system—Supplies process steam for sterilization of manufacturing equipment and processes.
High Efficiency Particulate Air Filter (HEPA) filtration of the room air supply—The efficiency of this air filter retains particles with a minimum size of 0.3 µm and has a particle retaining efficiency of 99.97%.
Washing and sterilization in a dry heat tunnel where glass components such as vials, syringes, and bottles are cleaned and sterilized by heat, normally in excess of 300°C
Processing of stoppers and other “rubber”-type components—Washing, sterilizing, depyrogenating, and sometimes siliconization are used to enhance component handling.
Steam autoclaves are used for sterilization of product contact parts (e.g., stoppers) and product flow parts (e.g., sterile filters, piping, valves, and filling machine parts). Most terminal sterilization is done in steam autoclaves, but gamma radiation is another good method of terminal sterilization.
Other types of sterilization technologies may be used (e.g., hydrogen peroxide, chlorine dioxide, ethylene oxide, and electron beam radiation).
Freeze drying—This is the process of removing water from a liquid product by sublimation to form a dried cake product that enhances stability. Product can be reconstituted by injecting sterile water.
Sterile filtration—Process of filtering a product through a 0.2-µm or smaller filter into a sterilized receiving vessel. The filtration process effectively removes the bacteria. Pyrogens or endotoxin must be controlled by cleaning the equipment and assuring that all drugs and chemicals are pyrogen free.
Restricted access barrier systems—A restricted access barrier systems filling line and associated transfers must be placed in a Grade A [International Organization for Standardization (ISO) 5] environment. The advantage of this approach is reduced cost and versatility of the production line.
Isolators require Grade C (ISO 8)—This is a more intense system that meets a higher standard than the restricted access barrier systems. The advantage of this approach is added assurance of control and sterility because the entire chamber is cleaned and sterilized. Another significant advantage is that employees are working in a Grade C environment. The disadvantages are that the set up for multiple products is more difficult and time consuming when using different components and the costs are greater.
High voltage and low current are effective to test for leaks from ampoules and vials with stoppers. Current flow across the vial is an indicator of a leak, and when found, the product is rejected. This technology needs access to the product to conduct the test
Determination of air flow when vacuum is applied to the container, which is in a sealed cupping device. There is no impact on the product.
Spark testing of freeze-dried products filled under full vacuum. This technology needs access to the product to run the test.
Vial head space gas analysis. This technology can determine leaks in a product that has been freeze dried and sealed with nitrogen in the head space. The testing, using laser technology, can check for oxygen and other gases in the vial head space without any impact on the product.
products made are covered by GMP as well as the return and destruction of all unused and partially used containers of drugs. A director is responsible for the overall GMP in terms of the product, processes, and controls. All GMP data must be quality assured by an audit, and the data must be archived per the company’s standard operating procedures.
Inability to use a common formulation. Having a single formulation worldwide is a goal that is often abridged because of different regulations and marketing practices in various countries. While many different formulations can theoretically be made in one factory, it is usually unfeasible because of the possibility of higher manufacturing costs, limited capacity, limited human resources, transportation costs, and regulatory considerations.
Need for a backup plant exists in case of a major problem such as fire, flood, prolonged strike, sabotage, and so forth. A company’s income could be seriously affected if any of those situations occur and affect a major source of company revenue. For investigational drugs, any of the problems mentioned could greatly delay development.
There are differences in a company’s production plants, techniques, and equipment in different countries that limit a company’s ability to use only a single plant.
Policies of the company to protect itself in case of problems also play a role in influencing this issue.
to make a new drug and it is uncertain whether a company desires to bring that technology in-house. A company may have a tradition of manufacturing all of their own products, but it makes no sense to rigidly adhere to this principle when it is economically preferable to have other manufacturers make a new drug. However, a thorough evaluation must be undertaken to understand the actual benefits and negative aspects of manufacturing in-house versus outsourcing to a contract manufacturer.
It is easy to define the cost of manufacturing.
The price is set by the contract, but be aware that there may be hidden costs.
It may serve as a backup for disaster planning.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


