Pheochromocytoma/Paraganglioma
Vania Nosé, MD, PhD
Arthur S. Tischler, MD
Key Facts
Terminology
Use of term pheochromocytoma restricted to adrenal medulla
Malignancy is defined by documentation of metastases to sites where normal paraganglia are not present
Etiology/Pathogenesis
At least 30% of PCCs are hereditary; at least 10 susceptibility genes are now known
Most attributable to mutations in RET, VHL, NF1, SDHA, SDHB, SDHC, SDHD, SDHAF2, KIF1B, TMEM127, and MAX
SDHx mutations account for up to 80% of familial PCC/PGL aggregations, 30% of pediatric tumors, and ˜ 50% of malignant tumors
SDHB mutation associated with extraadrenal abdominal location, high probability of metastasis, and poor prognosis
Clinical Issues
Identification of patients with hereditary PCC involves clinical assessment, biochemical testing, and pathology leading to directed genetic testing
Microscopic Pathology
Classic pattern is small nests (zellballen) of neuroendocrine cells with interspersed small blood vessels
Ancillary Tests
Immunohistochemistry for SDHB and SDHA can triage patients for appropriate genetic testing
 The typical pheochromocytoma has a gray-pink cut surface with areas of hemorrhage, which distinguishes it from the yellow-brown of adrenal cortex  . . |
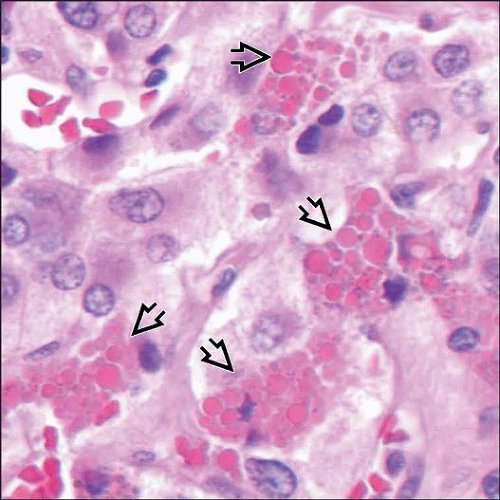 Pheochromocytomas in patients with multiple endocrine neoplasia type 2 (MEN2) syndromes may have numerous hyaline globules  that are particularly conspicuous in pheochromocytomas of these patients. that are particularly conspicuous in pheochromocytomas of these patients. |
TERMINOLOGY
Abbreviations
Pheochromocytoma (PCC)
Paraganglioma (PGL)
Definitions
Normal paraganglia consist of neural crest-derived neuroendocrine cells associated with sympathetic and parasympathetic nerves
Adrenal medulla and organ of Zuckerkandl are major sympathetic paraganglia; others are microscopic
Carotid bodies are major parasympathetic paraganglia; others are microscopic
PCC and PGL are catecholamine-secreting tumors of neural crest origin that arise from the adrenal medulla or extraadrenal sympathetic paraganglia, respectively
World Health Organization definitions of 2004 arbitrarily established terminology for tumors of paraganglia to eliminate previous inconsistent usage
PCC: Neuroendocrine tumor arising from chromaffin cells of adrenal medulla
Similar tumors in other locations are extraadrenal PGL (often abbreviated in practice to just PGL)
Sympathetic (sympathoadrenal) PGLs arise in vicinity of sympathetic chains and along sympathetic nerve branches in pelvic organs and retroperitoneum
Parasympathetic PGL (a.k.a. head and neck PGL [HNP]) arise mainly from branches of vagus and glossopharyngeal nerves in head and neck, sometimes mediastinum
PCC is an intraadrenal sympathetic PGL
ETIOLOGY/PATHOGENESIS
Hereditary PCC/PGL
Over the last decade, extensive genetic heterogeneity of these tumors came to light with identification of multiple susceptibility genes
Most striking feature is genetic diversity
≥ 1/3 of PCCs/PGLs are hereditary
These mutations account for ≥ 1/3 of PCC and PGL
Highest inheritable proportion of any known human tumor
Occult germline mutations of susceptibility genes common in patients with apparently sporadic tumors
≥ 10 susceptibility genes now established
Most attributable to mutations in RET, VHL, NF1, SDHA, SDHB, SDHC, SDHD, SDHAF2, KIF1B, TMEM127, and MAX
Most recently identified hereditary forms of PCC and PGL are SDHx, transmembrane-encoding gene, TMEM127, and MYC-binding partner, MAX
Greater understanding of molecular signals transduced by these genes and their respective mutants has advanced our understanding of kinase signaling pathways, hypoxia regulation, and link between metabolic disruptions and cell growth
Multiple endocrine neoplasia type 2 (MEN2)
Autosomal dominant syndrome caused by mutation of RET proto-oncogene
Activating RET mutations predispose to PCCs, which are often recurrent and bilateral but typically have a low risk of malignancy
PCC are bilateral in 50-80% of cases but are almost always benign
Familial PGL/PCC syndromes
PGL syndromes encompass a group of inherited syndromes which involve mutations in the genes that encode components of the succinate
dehydrogenase (SDH) mitochondrial enzyme complex 2
SDH is composed of 4 subunits: A, B, C, and D
Germline mutations in SDHx genes give rise to familial PCC/PGL syndrome, sometimes only referred to as familial PGL
Associated with germline mutations in genes encoding subunits of SDH enzyme complex in context of familial PGL syndromes
PGL1, PGL 2, PGL3, and PGL4 caused by mutations in the SDHD, SDHAF2, SDHC, and SDHB genes, respectively
Familial PGL syndrome, PGL2, is caused by mutations in SDHAF2/SDH5, which encodes for a molecule that is an accessory to the function of the SDH enzyme and its SDHA subunit
SDHA-related PGLs are rare and are caused by loss-of-function mutation in SDHA
Carney triad
Mean age of presentation of PGL/PCC: 28 years
Only 16% present with PCC
von Hippel-Lindau syndrome (VHL)
Autosomal dominant disorder caused by mutation of VHL
About 10-26% of VHL patients develop PCC or PGL, but risk varies between different families
Frequency of PCC in individuals with VHL is 10-20%
Mean age of onset of PCC in VHL: ˜ 30 years
Neurofibromatosis type 1 (NF1)
Autosomal dominant disorder caused by mutation of NF1
PCCs occur in 20-50% of individuals with NF1 and hypertension
NF1-associated PCCs and PGLs typically have characteristics similar to those of sporadic tumors, with a relatively late mean age of onset and about 10% risk of malignancy
Gangliocytic duodenal PGL may occur in patients with NF1
Approximately 84% of PCC are unilateral
Carney-Stratakis dyad
Inherited predisposition to gastrointestinal stromal tumor (GIST) and PGL caused by inactivating germline mutations in SDHB, SDHC, or SDHD
Only rare cases reported to be associated with PCC
Most recently identified hereditary forms of PCC and PGL are transmembrane-encoding gene, TMEM127, and MYC-binding partner, MAX
So far, no specific syndrome has been described for TMEM127
MAX mutations occur in families with PCC, but no specific syndrome has been described yet
Sporadic PCC/PGL
Majority of PCCs appear to arise sporadically
Only occasionally harbor somatic mutations except for NF1, which is mutated in > 25% of sporadic tumors
Germline mutations in known susceptibility genes may be seen in up to 16% of sporadic-appearing cases
Changes in copy number of hereditary susceptibility genes may be present
Environmental Influences
High-altitude PGL in people and cattle living in mountainous areas of some countries
Mostly carotid PGL
CLINICAL ISSUES
Site
˜ 98% of sympathetic PGLs are located in abdomen or pelvis; 90% are adrenal PCCs
Most parasympathetic PGLs are carotid, jugulotympanic, or vagal
Presentation
Depends on tumor location
Sympathoadrenal PCCs/PGLs usually cause signs and symptoms of catecholamine excess
Tumors with SDHB gene mutation are more likely than other sympathoadrenal PCCs/PGLs to be clinically silent
Parasympathetic PGLs are usually clinically silent mass lesions
Hereditary PCC/PGL often found after other stigmata point to hereditary tumor syndrome (usually MEN2, VHL, NF1)
Gastrointestinal stromal tumors are important component of several new syndromes with mutated SDHx
Variants of some hereditary syndromes can cause only PCC/PGL (VHL type 2C)
Mutations of some genes (e.g., TMEM127) cause hereditary but nonsyndromic PCC/PGL (no associated abnormalities)
Affected by genotype
Sporadic tumors solitary, usually in adults
Multiple tumors or tumors presenting in children suggest hereditary disease
Tumors with RET or NF1 mutations almost always intraadrenal
Abdominal PGL or combination of sympathetic and parasympathetic PCC/PGL suggests SDHx mutation
SDHD– and SDHAF2-related PGL show parent-of-origin dependent expression; tumor development only with paternal inheritance
Identification of patients with hereditary PCC/PGL involves clinical assessment, biochemical testing, and pathology leading to directed genetic testing
Laboratory Tests
Plasma metanephrine and normetanephrine more sensitive than corresponding catecholamines for tumor detection
Methoxytyramine: New marker sometimes produced by clinically nonfunctional tumors, especially with SDH mutations
PCC can be adrenergic or noradrenergic; extraadrenal PGL almost always noradrenergic; HNP can lack ability for catecholamine biosynthesis
Genotype affects biochemical function
Noradrenergic PCC raises suspicion of VHL
Treatment
Complete surgical excision is only cure
Unresectable primary tumors and metastases can have long doubling time; watchful waiting often a viable option
Prognosis
Most patients with metastases eventually die from complications of excess catecholamines or destructive local growth
Malignancy
World Health Organization 2004 defines malignancy by presence of metastasis
Must be to sites where normal paraganglia are not present to avoid confusion with new primary tumor
Currently, no generally accepted histological criteria to predict whether primary PCC or PGL will metastasize
Extensive local invasion alone is poor predictor of metastasis
Predictive value of tumor size is controversial
Risk of metastasis and prognosis vary with tumor location and genotype
˜ 10% metastasis for PCCs, > 20% for PGLs
Best predictor of metastasis is presence of SDHB mutation (> 30%)
After metastases occur, worst prognosis is for tumors caused by SDHB mutation
Metastases can develop years or decades after resection of primary tumor
Currently recommended that no PCC/PGL be signed out as benign; all patients receive lifelong follow-up
IMAGE FINDINGS
General Features
Anatomic imaging
MR: Very intense T2-weighted image (light bulb sign) is classic but not always present
Contrast-enhanced CT
Functional imaging
More specific because based on specific aspects of tumor phenotype
More sensitive for small tumors or metastases in bone
Efficacy of different functional imaging techniques varies according to tumor genotype
MACROSCOPIC FEATURES
General Features
Cut surface usually pink-gray to tan, distinguishes PCCs from yellow-gold of most adrenal cortical tumors
Occasional tumors show patchy or diffuse brown pigmentation
Cystic degeneration and necrosis sometimes present
Medullary hyperplasia, when present, may indicate hereditary form of the disease
MICROSCOPIC PATHOLOGY
Histologic Features
Classic pattern is small nests (zellballen) of neuroendocrine cells (chief cells) with interspersed small blood vessels
Numerous variant and combined patterns exist, including diffuse growth, large zellballen, spindle cells, cell cords
Sustentacular cells variably present, best seen with IHC
Possibly nonneoplastic cell type induced or attracted by tumor-derived factors
Cavernous blood vessels sometimes prominent, especially in HNP



