Introduction
Following preclinical and first-in-human (Phase I) studies, an investigational drug or device is studied in subjects who have the disorder of interest. This phase of development is commonly called Phase II or exploratory therapeutic development. Major goals of Phase II studies are to support the proof of concept or proof of mechanism identified during preclinical development and to show evidence of efficacy that will support future confirmatory trials (Table 7–1). Other objectives include defining the target population, exploring the pharmacodynamic relationship between dosing regimens and the effects on disease, determining the proposed dosing regimen for future trials, and providing preliminary estimates of drug effect to be used in calculating sample sizes for Phase III trials.
|
The objectives of Phase II support a critical “go/no-go” decision with regard to further clinical development. Sponsors usually prefer to abandon ineffective drugs early rather than waiting for resource- and time-intensive Phase III trials to tell them that the therapeutic has failed. Proof of mechanism, proof of concept, and identification of a therapeutic dose range and regimen are desired as early as possible. An inability to define any of these delays the clinical development program and may ultimately “kill” the therapeutic. Thus, the Phase II program should support the predefined goals of the clinical development plan and the target product labeling. The critical decision points and the criteria for the go/no-go decision are often determined as part of the clinical development plan.
Dividing the clinical drug development process into numbered phases implies a linear progression; in fact, Phase II activities regularly occur while pivotal Phase III trials are active. Such overlap with other phases is not uncommon, particularly when a drug is evaluated in understudied subgroups within a target population. In some cases, a well-designed Phase II dose–response study can serve as one of the pivotal studies to be submitted to regulatory agencies for marketing approval.
Characteristics of Phase II Studies
Phase II trials are generally short, enroll a relatively small number of subjects (up to 300), and often use surrogate rather than clinical end points (1,2). Phase II is sometimes divided into Phase IIa and Phase IIb (or early and late Phase II) to indicate the shifting goals of this phase over time. Phase I and IIa activities collectively represent “completion of clinical trials that provide data on the relationship of dosing and response for the particular intended use (including trials on the impact of dose ranging on safety, biomarkers, and proof of concept)” (3). In contrast to Phase IIa trials, late exploratory therapeutic trials tend to last longer, have larger sample sizes, and place greater emphasis on the clinical efficacy end points to be used in Phase III.
Phase II seeks to define the optimum loading and maintenance doses, dosing interval, and treatment duration in humans. Investigators commonly give multiple doses of the study drug against a comparator or placebo, and a dose–response curve is generated that describes the pharmacodynamic relationship between the dosing regimen and effect. The shape of the dose–response curve, tolerability at varying doses, maximum tolerated dose, minimum dose achieving maximum desired effect, and the safety and efficacy of varying drug doses are studied. The study drug also can be given with commonly coadministered medications to explore drug–drug interactions. Information from studies of other compounds with a similar mechanism of action can be useful—for example, to help select a surrogate end point or a study population.
The drug doses incorporated into early Phase II trials are determined from preclinical safety and effectiveness data and Phase I pharmacokinetic studies. A parallel-group design with several fixed doses is common (Figure 7–1). There are often several treatment groups with different dosing regimens, and each enrolled subject is assigned to one of the treatment groups. Late Phase II trials select dose regimens that appear optimal from earlier studies, but they can also include additional exploratory dosing schemes.
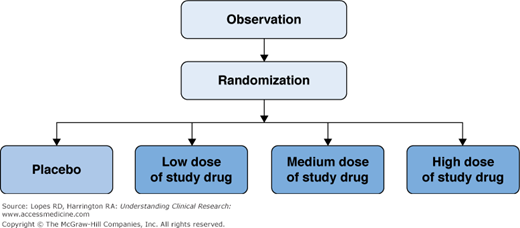
In some cases, an adaptive study design is used (4), in which scheduled interim data analyses drive predetermined changes in the trial’s conduct, such as shifting enrollment to treatment subgroups that are more promising, or eliminating treatment groups (e.g., a failing dose arm) altogether. To improve the chances of success in Phase III, a potentially safe and effective dose regimen in the appropriate patient population must be identified during Phase II with as much certainty as possible.
Additional studies might be required later during development to support the use of the study drug in subpopulations such as elderly persons or patients with renal dysfunction or hepatic impairment. Emphasis has increased recently on including special populations earlier in clinical development (e.g., during Phase II) (5,6). It also might be relevant to evaluate drug dosing in various stages of a disease, such as early after disease onset versus late stage, and in patients with different disease severities.
Interest is increasing regarding the use of drug modeling and simulation to assist in dose finding. In this context, Phase I and II pharmacokinetic and pharmacodynamic data are used to simulate dosing in humans, hopefully leading to earlier identification of an effective dosing strategy and reduced development time.
Effect of the Globalization of Drug Development on Phase II
Although in the past the pharmaceutical industry relied on a small number of domestic sites for Phase II studies, drug development has increasingly become a global enterprise. More clinical trials are being conducted outside the United States, and pharmaceutical companies might outsource many roles within a study to international contract research organizations (CROs) or academic research organizations (AROs) (7). Studying drugs outside the United States may confer several advantages to manufacturers, including reduced costs, faster enrollment, and fewer local regulatory hurdles. In addition, drug metabolism and drug effects can differ by region and genetic makeup; if drugs are to be marketed in diverse populations, it is often necessary to study them in different geographic regions or in different genetic populations. These factors, together with potentially large increases in markets in developing countries, have led to more Phase II studies being conducted outside the United States. Even small Phase II trials now commonly involve multiple centers and international collaborators. Despite the potential rewards, this trend increases the complexity of trials in many areas, including regulatory agency interactions, institutional review boards (IRBs), site monitoring, data management, and ensuring data quality, drug supply, safety monitoring, and communication among investigators, trial sponsors, contractors, etc. As an example, an international Phase II study that includes multiple timed blood sampling for pharmacokinetic measurements may require detailed, multilingual instructions for obtaining the samples, as well as complicated logistics for shipping them to a central laboratory in another country for processing and storage until the assays are completed. An in-depth discussion of international research can be found in Chapter 4.



