Introduction
The development process for a new molecular entity (NME) in humans proceeds over several phases of clinical trials (I through III) before it can be approved for marketing by national regulatory authorities (Figure 6–1). Once regulatory approval has been obtained, after undergoing all required trials over many years, NMEs translate into drugs that are used to treat specific conditions.
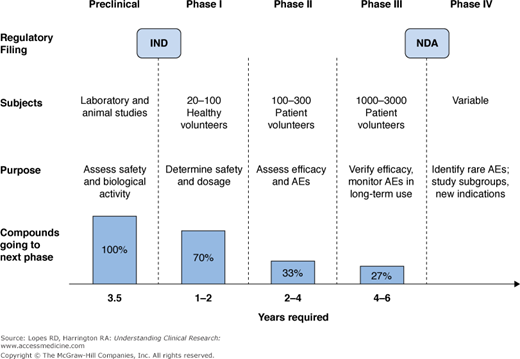
The first step in human drug development involves Phase I or “first-in-human” studies. These trials are an exciting milestone in the drug development process, because this is the first time an investigational drug is given to humans. Before human testing occurs, preclinical (animal and laboratory) work establishes the preliminary safety profile of the NME and a starting dose for use in humans. At the end of the preclinical phase in the United States, the drug’s developer submits an Investigational New Drug application (IND) to the US Food and Drug Administration (FDA) to obtain permission to begin human testing of the compound. The performance of an NME in Phase I trials helps determine its progression through the drug development process.
The primary goal of Phase I trials is to assess the safety of the NME in human subjects. In addition, the NME’s pharmacokinetics (“what the body does to the drug”) are evaluated to determine the most appropriate dose and dosing regimen in humans. Phase I studies also can explore the effects of different formulations, different exposure routes, giving the drug under fasting or fed conditions, and drug–drug interactions. Because safety and pharmacokinetics are primary objectives of Phase I trials, they are usually conducted in healthy volunteers (often adult men). This strategy limits factors that can confound assessments of safety and pharmacokinetics, which are often present in patients with the disease of interest. However, in the case of significant toxicity concerns or a narrow therapeutic index of the NME, these trials may be conducted in actual patient populations.
The design of Phase I clinical trials varies according to the primary objective and can range from open-label to blinded, randomized trials. Phase I trials must show that an investigational agent is safe enough to be used in humans and must generate enough pharmacokinetic data to permit the design and conduct of Phase II trials. Description of pharmacodynamics (“what the drug does to the body”) and early tests of efficacy can be secondary end points of Phase I trials, but the emphasis is on nontherapeutic objectives.
Phase I studies present several practical challenges to drug manufacturers. For example, the compound might not be available yet in the optimal formulation for the patient population of interest. Manufacturing the compound for these studies also can be very expensive because production of the NME has not been scaled up. Companies must balance the likelihood of the drug succeeding versus the resources required for the full-scale manufacturing process.
Required Preclinical Safety Evaluations
During the 1980s, scientists at DuPont began investigating a new class of antihypertensive agents that worked by antagonizing (blocking) the angiotensin II receptor. Early in vitro tests showed that adding angiotensin II to arterial tissue caused contraction of the artery, but adding angiotensin II and an experimental angiotensin II antagonist prevented contraction. After extensive engineering of the molecule to increase potency and obtain an orally active formulation, this candidate drug, dubbed DuP 753, was tested in rats and dogs (1–3). It was safe and efficacious in both species, but metabolism in rats created a highly active metabolite that increased the candidate drug’s potency and prolonged its half-life. This metabolite was not created in dogs.
Before undergoing Phase I trials, an NME must go through extensive preclinical evaluations. These are done primarily in animal models that closely approximate the disease and the expected impact of the NME in humans. Animal models offer opportunities to elucidate drug mechanisms of action that would not be possible in human studies. For example, investigators studying DuP 753 hypothesized that the NME lowered the blood pressure by inhibiting the renin system. In one preclinical experiment, investigators removed both kidneys from anesthetized rats and noted that without the renin produced by the kidneys, DuP 753 did not reduce blood pressure (1). This finding confirmed the hypothesized mechanism of action of DuP 753; such a study could not have been ethically done in humans. Another important pharmacokinetic finding from preclinical studies of this NME was that production of a pharmacologically active metabolite was species dependent, occurring in rats but not in dogs (4,5). This finding raised the question of whether humans would produce such a metabolite, an important objective to define during Phase I human clinical testing. The presence of active metabolites could have safety, dosing, and efficacy implications.
Below are listed some of the preclinical studies that must be completed before an agent can be given to humans.
- Safety pharmacology in animals assessing the impact of the drug on vital functions (such as the cardiovascular system).
- Animal pharmacokinetic assessments, including evaluation of drug absorption, distribution, metabolism, and excretion (ADME).
- Single-dose (or dose-escalation) toxicity studies in two animal species.
- Repeated dose–toxicity studies in two animal species (one nonrodent) with duration as long as or longer than the proposed Phase I studies in humans. Additionally, the route of administration tested (oral, intravenous) should be the same as the proposed route of administration for humans.
- In vitro genotoxicity studies to assess mutagenicity.
- Reproductive toxicity studies to assess the impact of a drug on fetal development and female fertility, although this varies by region (5a):
- United States—embryo-fetal development and female fertility studies do not have to be done before Phase I trials in women of childbearing potential, if the female subjects are carefully monitored, using appropriate contraception, and taking precautions to minimize risk of pregnancy.
- European Union—a drug’s impact on embryo-fetal development must be assessed before giving the drug to women of childbearing age. Studies assessing the impact on female fertility must be done before Phase III trials.
- Japan—embryo-fetal development and female fertility studies must be done before giving the drug to women of childbearing age. These women must also be using appropriate contraception.
- United States—embryo-fetal development and female fertility studies do not have to be done before Phase I trials in women of childbearing potential, if the female subjects are carefully monitored, using appropriate contraception, and taking precautions to minimize risk of pregnancy.
Determination of the Maximum Recommended Starting Dose
The animal data showed DuP 753 to be a highly selective angiotensin II receptor antagonist. This agent, if brought to market, would be competing against angiotensin-converting enzyme (ACE) inhibitors. ACE inhibitors are not selective for ACE. They also act on the bradykinin and substance P systems, resulting in side effects, such as dry cough and angioneurotic edema, that often limit their tolerability. If DuP 753 was more selective, these side effects could be minimized, offering DuP 753 a competitive advantage over ACE inhibitors. Based on the selectivity of DuP 753 and its safety profile in animals, investigators established a maximum recommended starting dose (MRSD) of 40 mg (6).
Because the MRSD will be the dose tested in Phase I trials, it must be chosen carefully considering safety and trial efficiency. A conservative approach to implementing MRSD in humans (choosing a low dose) might lead to prolonged and expensive Phase I trials, testing several dosing levels to determine the most appropriate one. In contrast, an aggressive approach (choosing a high dose) can lead to excess toxicity. The MRSD is often extrapolated from preclinical data collected from animal models (Figure 6–2) (7).
Figure 6–2.

Example of allometric scaling of animal dose to human dose. Reproduced, with permission, from Peterson JK, Houghton PJ. Integrating pharmacology and in vivo cancer models in preclinical and clinical drug development. Eur J Cancer. 2004;40(6):837-844. CLtot, total clearance; MTD, maximum tolerated dose; TD, tolerated dose; W, weight.
An FDA Guidance for Industry (8) outlines an algorithm for estimating the MRSD in initial trials in adult healthy volunteers (Figure 6–3). The algorithm follows these steps:
No observed adverse effect levels (NOAELs) are extrapolated from data obtained from the most appropriate animal model. The NOAEL is determined from preclinical toxicology studies assessing overt toxicity, surrogate markers of toxicity, and exaggerated pharmacodynamic effects. The most appropriate animal model is usually the one most sensitive to the drug, but other factors can influence this choice, including variations in ADME among species and evidence that a particular animal model is most predictive of human toxicity.
NOAELs are then normalized and scaled to human-equivalent doses (HEDs) using body surface area-based allometry. The FDA has derived conversion factors for different species by which NOAELs can be multiplied to determine HEDs (Table 6–1) (8).
Extrapolation of animal data to humans is imperfect, so, after the HED is calculated, a safety factor is applied to incorporate a margin of error into the MRSD. Conventionally, the MRSD is calculated by dividing the HED by a safety factor of 10. In cases of severe or irreversible toxicity, narrow therapeutic index, novel therapeutic targets, or an animal model with limited ADME applicability to humans, a safety factor >10 may be applied. Conversely, a safety factor <10 might be applied for an NME that is a member of a well-described drug class with similar metabolism, toxicity, route, and frequency of administration. Drugs with limited, easily monitored, and reversible toxicity also might warrant a safety factor <10.
Figure 6–3.
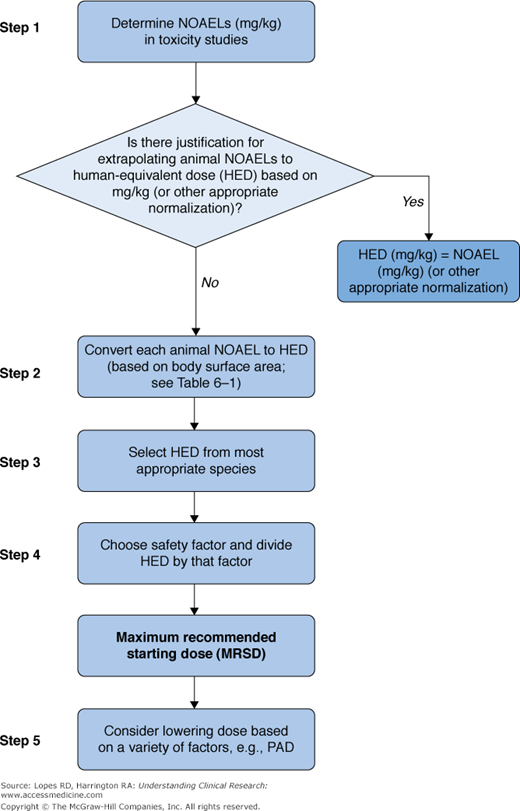
Selection of maximum recommended starting dose for drugs administered systemically to normal volunteers. NOAELs, no observed adverse effect (drug) levels; PAD, peripheral arterial disease. Reprinted from US Food and Drug Administration. Guidance for Industry on Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; Availability. 2005. . Accessed April 11, 2012.
To convert animal dose (mg/kg) to HEDa (mg/kg) either | |||
|---|---|---|---|
Species | To convert animal dose (mg/kg) to dose in mg/m2, multiply by km | Divide animal dose by | Multiply animal dose by |
Human | 37 | — | — |
Child (20 kg)b | 25 | — | — |
Mouse | 3 | 12.3 | 0.08 |
Hamster | 5 | 7.4 | 0.13 |
Rat | 6 | 6.2 | 0.16 |
Ferret | 7 | 5.3 | 0.19 |
Guinea pig | 8 | 4.6 | 0.22 |
Rabbit | 12 | 3.1 | 0.32 |



