Pharmacodynamics and Pharmacokinetics
General Principles
The effect of any medication is determined by three variables:
Affinity for and intrinsic activity at a site of action (pharmacodynamics, variable 1)
Drug concentration (pharmacokinetics, variable 2)
The patient’s biological state (variable 3)
Pharmacodynamics describes what a drug does to the body (good or bad). For example, an antipsychotic that blocks the dopamine-2 (DA2) receptor is capable of reducing certain symptoms (e.g., hallucinations) but may also cause extrapyramidal side effects (e.g., dystonia) (1,2 and 3).
A sufficient amount of the drug must reach the site of action under clinically relevant dosing conditions to have a physiologically meaningful effect. This is the domain of pharmacokinetics (4,5,6 and 7).
Finally, various biological factors specific to a given patient can impact a drug’s ultimate effect.
Pharmacodynamics (Variable 1)
This variable considers what a drug does once it interacts at adequate concentrations with its biological target (e.g., receptor). In this context, two critical factors are the affinity (or strength) for a drug’s attachment to a target and its intrinsic activity (e.g., full or partial agonist, antagonist, or inverse agonist at a receptor). Psychotropic drugs affect specific biochemical processes, most often involving enzymes, receptors, or ion channels. A given drug’s action that produces a physiological response (whether intended or otherwise) is termed the mechanism of action.
Perhaps, most critical to a discussion on pharmacodynamics is the explosion of knowledge about the fundamental subcellular processes that contribute to psychiatric disorders and can be impacted by the effects of psychotropics. In this context, we have been able to isolate and study many biologically important substrates (e.g., neurotransmitters, receptor subtypes, and various components of the postligand-receptor interface, including various subsequent messenger systems). The goal is to characterize the cascade of intraneuronal events that lead to genetically determined cell protein alterations. Once these processes are clarified, they can provide insights into pathoetiology, as well as become targets for specific drug development. Concurrently, observing how new drugs interact with these cellular components enhances our knowledge of their functional role in mediating specific behavioral symptoms. Newer agents can then serve as probes to test whether these components are relevant to a given psychiatric disorder.
It is axiomatic that central effects mediate the clinical actions of psychotropic medications. But, for any given drug, the effects on known processes may not be the mechanism mediating clinical response. Instead, the clinical outcome may be the result of an unrecognized central action. This is due to our limited understanding of the pathophysiology underlying specific psychiatric disorders. Thus, when the fundamental biology underlying a disorder is unknown, it is not possible to state how a drug is correcting a given syndrome.
With the rapid developments in neuroscience, we will increase our reliance on drug probes to test the functional integrity of neurotransmitter systems in specific disorders. The ever-increasing number of biochemical processes that may mediate the effects of a specific psychotropic include
Enzymes responsible for the synthesis and degradation of an expanding list that includes neurotransmitters, neuropeptides, and neurohormones
Storage of neurotransmitters in vesicles within the cytoplasm
Release mechanisms
Presynaptic membrane uptake pumps
Subtypes of pre- and postsynaptic receptors
Ion channels
Receptor subcomponents (e.g., G-proteins)
Subsequent messenger systems (cyclic adenosine monophosphate [cAMP], protein kinase C [PKC]) (8)
Enhancement of neurotrophic or neuroprotective factors (e.g., brain-derived neurotrophic factor [BDNF])
Genetic transcription processes in the cell nucleus
As they become better characterized, these processes will increasingly serve as targets for future drug development.
DRUG DEVELOPMENT AND ITS IMPLICATIONS
The first psychotropics of the modern era (e.g., lithium, first-generation antipsychotics [FGAs], tricyclic, and monoamine oxidase inhibitor antidepressants) were discovered serendipitously. These agents were not engineered to have selective actions but produce a wide range of central biochemical effects. Thus, they usually affect more than one neurotransmitter system simultaneously, resulting in multiple repercussions, including
They may help in more than one condition because they act by more than one mechanism.
Any number of their actions could be responsible for the intended clinical effect; therefore, such drugs provide limited insight into the pathophysiology of a given condition.
Generally, due to their multiplicity of effects (positive as well as negative), these “broad-spectrum” medications are less well tolerated than agents with fewer biochemical interactions.
Future attempts to focus the actions of newer agents should reduce their “signal-to-noise ratio” and improve specificity (9).
Although there are many problems with firstgeneration psychotropics, they are effective and provide some insights into underlying pathophysiology. Studying the effects of these agents on specific biochemical functions has heuristic value, generating numerous hypotheses that guide subsequent drug development. One set of hypotheses deals with the possible mechanisms underlying the clinical efficacy of these agents, including
The dopamine hypothesis, based on the actions of FGAs
The catecholamine and the indolamine hypotheses, based on the actions of various antidepressants
The permissive, adrenergic-cholinergic balance, and bidimensional hypotheses, based on both the effects of antidepressants and on the modulating interactions among various neurotransmitter systems
Concurrently, hypotheses were developed regarding the undesirable or toxic effects associated with the various biochemical actions of these agents. Examples include
Orthostatic hypotension secondary to α-adrenergic receptor blockade
Cardiotoxicity secondary to membrane stabilization
Central anticholinergic syndrome due to the potent muscarinic-cholinergic effects of many psychotropics
Chemists can better define the structureactivity relationship of early psychotropics to guide the development of newer drugs. Such relationships are refined by in vitro testing to determine whether newly synthesized compounds have the desired biochemical effect on specific targets, such as enzyme inhibition or receptor blockade. Simultaneously, these agents are also tested for any undesirable effects. Modifications can then be made to the chemical structure to eliminate or reduce such unwanted qualities. When a new psychotropic drug meets the desired “inclusion and exclusion criteria,”
it is tested clinically to determine whether it possesses the desired therapeutic effect. Results from these clinical studies provide critical feedback regarding the mechanisms of action, guiding the development of future agents.
it is tested clinically to determine whether it possesses the desired therapeutic effect. Results from these clinical studies provide critical feedback regarding the mechanisms of action, guiding the development of future agents.
For example, the selective serotonin reuptake inhibitors (SSRIs), bupropion, venlafaxine, nefazodone, mirtazapine, and duloxetine represent agents engineered to eliminate many of the earlier-generation antidepressant adverse effects (10). Beginning in the late 1980s, the introduction of these agents had important research and clinical implications. From the research standpoint, they advanced our understanding of the pathophysiology of various disorders, while providing a way to test for the existence of putative biochemically distinct subtypes. This was not possible with earlier-generation psychotropics, due to their lack of specific action. For example, the tricyclic antidepressants (TCAs) are effective in depressive disorders, enuresis, and panic attacks, but different mechanisms are believed to be responsible for these varied efficacies. In a similar way, all TCAs have effects on both the norepinephrine and the serotonin reuptake systems within the clinically relevant concentration range. Attempts in the past to distinguish between “serotonergic” and “adrenergic” depressive disorders with such agents were unsuccessful because of the nonspecific nature of the effects of these drugs, even if two such forms of depression exist. Since newer agents are more specific in their affinity for one neurotransmitter system versus another, they may help to expand our understanding of the relevant neurobiology. Clinically, these agents also have several advantages because they
Have more specific pharmacological actions
Can test the functional integrity of a given neurotransmitter system in a specific syndrome or patient
Are generally safer and better tolerated
Thus, the first era of modern psychopharmacotherapy (i.e., serendipitous discovery) is giving way to the second era (i.e., refinement of drugs based on known biochemical effects). This process will eventually lead to the next era, which will be the synthesis of compounds with specific interactions at newly discovered subcomponents of the neuron. The existence of such agents may also permit the development of an empirically based hierarchical treatment plan that defines the agent of first choice as well as agents likely to help when the first choice is unsuccessful or poorly tolerated. The approach used in this book allows the reader to both anticipate such developments and incorporate them as they occur.
Pharmacokinetics (Variable 2)
Drug concentration determines which site(s) of action a drug will engage and to what degree. Concentration is determined by a drug’s pharmacokinetics, or what the body does to the drug. Pharmacokinetics literally refers to drug movement, which is divided into four phases:
Absorption of the drug from the site of administration
Distribution through the body
Metabolism or biotransformation into more polar metabolites
Elimination from the body
Drug concentration is determined by the dosing rate, as well as the clearance of the drug. Thus, clearance is as important as the dosing rate to determine the ultimate concentration of a drug.
Pharmacokinetics is also relevant to the potential utility of therapeutic drug monitoring (TDM; discussed later in this chapter). For most drugs, an adequate occupation of the targeted site of action is needed to achieve an optimal response. Below a critical level, the target will not be sufficiently engaged to produce a physiologically meaningful effect. Conversely, occupation above a critical upper limit is associated with an increased likelihood of adverse effects (2,3).
FOUR PRIMARY PHARMACOKINETIC PHASES
We will discuss each of the four phases of pharmacokinetics, beginning with a case vignette to illustrate their clinical relevance.
Absorption
Bioavailability refers to the portion of a drug absorbed from the site of administration. The reference site of administration is intravenous,
because this route produces 100% absorption. Figure 2-1 illustrates three sample drug concentration curves in plasma as a function of time. The area under the curve (AUC) is the total amount of drug in the systemic circulation available for distribution to the sites of action. The same dose completely absorbed from any of these routes would produce an identical AUC (i.e., 100% bioavailability), although the shape would differ.
because this route produces 100% absorption. Figure 2-1 illustrates three sample drug concentration curves in plasma as a function of time. The area under the curve (AUC) is the total amount of drug in the systemic circulation available for distribution to the sites of action. The same dose completely absorbed from any of these routes would produce an identical AUC (i.e., 100% bioavailability), although the shape would differ.
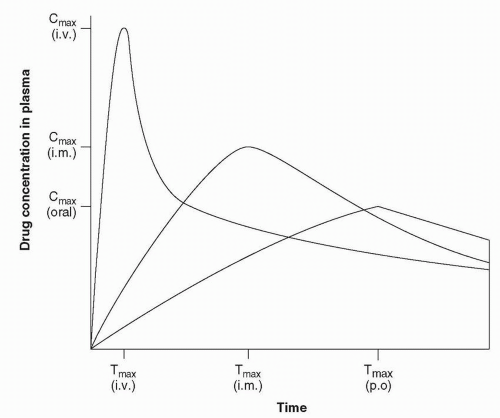 Figure 2-1 Single-dose plasma drug concentration versus time curves for the same dose of the same drug given to the same individual by three different routes. (From Janicak PG, Davis JM. Pharmacokinetics and drug interactions. In: Sadock BJ, Sadock V, eds. Kaplan & Sadock’s Comprehensive Textbook of Psychiatry, Vol. 2, 7th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2000:2251, with permission.) |
Case Example
A 37-year-old schizoaffective patient received thioridazine (100 mg by mouth four times daily), phenytoin (100 mg by mouth four times daily), and amitriptyline (50 mg by mouth four times daily). Although stabilized on this regimen for several weeks, the patient complained of daytime sedation. The treating physician then combined all three drugs into a single bedtime regimen. The patient expired the first night of the new schedule from an acute cardiac arrest.
In this case, each of the drugs was individually capable of slowing intracardiac conduction in a concentration-dependent manner. Death occurred as a result of their additive effect amplified by the decision to combine all three in a single nighttime dose. The increased peak concentration of each drug resulted in a fatal arrhythmia.
The principal route of administration for psychoactive drugs is oral, with absorption generally occurring in the small bowel. The drug is then absorbed into the portal circulation and enters the liver. Drugs can be metabolized by cytochrome P450 (CYP) enzymes in both the bowel wall and
the liver before reaching the systemic circulation (i.e., first-pass metabolism). Most psychiatric medications are highly lipophilic, so they readily pass the blood-brain barrier (BBB) and enter the central nervous system (CNS) (11,12,13 and 14). In addition, because of their high lipophilicity, they generally share several other features:
the liver before reaching the systemic circulation (i.e., first-pass metabolism). Most psychiatric medications are highly lipophilic, so they readily pass the blood-brain barrier (BBB) and enter the central nervous system (CNS) (11,12,13 and 14). In addition, because of their high lipophilicity, they generally share several other features:
Rapid and complete absorption
Rapid and extensive distribution in tissue compartments
High first-pass effect
Large volume of distribution
Any decrease in the total AUC for intramuscular or oral versus intravenous administration would represent a decrease in bioavailability based on that route of administration (15). Common factors that influence bioavailability include
Physicochemical properties of the drug
Formulation of the product
Disease states that influence gastrointestinal function or first-pass effect
Precipitation of a drug at the injection site
Drug interactions mediated by CYP isoenzymes in the bowel wall, liver, or transporters in the bowel wall
Drug interactions mediated by plasma glycoprotein (P-gp) in several organs including the capillary endothelial cells comprising the BBB
There are other clinically important parameters beyond the extent of absorption that are apparent on the single-dose plasma curve. These include
Peak concentration (Cmax)
Time to the peak concentration (Tmax)
Generally, Cmax will be inversely correlated to Tmax (i.e., the shorter the time for a drug to be absorbed, the higher the peak concentration). A higher Cmax and shorter Tmax typically mean a more rapid appearance of clinical activity after administration. These parameters can also determine whether a drug should be developed for a specific indication because Tmax and Cmax are generally a function of a compound’s physicochemical properties.
In terms of physicochemical properties, the more polar a compound, the slower the absorption from the gastrointestinal tract and the slower the penetration into the brain from the systemic circulation. These two phases (i.e., into the systemic circulation and into the brain) are usually correlated. Oxazepam, the most polar benzodiazepine (BZD), is slowly absorbed into the systemic circulation as well as into the brain (16,17). Such pharmacokinetics is usually not the profile desired in a sedative-hypnotic. In contrast, lorazepam rapidly penetrates both compartments, inducing sleep in a relatively rapid time frame (13). Although rate of penetration into the brain generally parallels rate of penetration into the systemic circulation, this is not always the case. An example is the original formulation of temazepam, which consisted of a hard gelatin capsule resistant to gastric breakdown (16). This formulation resulted in a slow absorption rate, diminishing its effectiveness even though it penetrated the brain rapidly from the systemic circulation. A change in the formulation led to more rapid absorption, increasing its usefulness as a sedative-hypnotic.
Fast absorption may not always be desirable, because adverse effects may be a function of Cmax. In the case example that began the discussion on absorption, failure to appreciate this fact led to a fatal outcome. Cardiac toxicity, due to the stabilization of excitable membranes, is as much a function of the peak plasma concentration as it is of the steady-state tissue concentration. Thus, changing a formulation to delay Tmax and reduce Cmax may significantly increase safety (e.g., reduced seizure risks with the two different sustained release formulations of bupropion). Dividing the dose into smaller amounts and administering it more frequently can also accomplish the same result. In such a case, the average plasma concentration over the dosing interval and the amount absorbed usually remains the same, but the peak concentration is lower and the trough concentration higher.
Differences in bioavailability, particularly as they affect the rate of absorption, can vary significantly among formulations (i.e., products) of the same drug. The U.S. Food and Drug Administration (FDA) considers a generic product to be comparable to a brand name if there is no more than a 20% difference (more or less) in bioavailability (i.e., Tmax and Cmax) (18). Theoretically, this could produce as much as a 40% difference between two generic preparations of the same drug. This possibility may explain why a patient who previously tolerated and benefited from one generic preparation of a medication begins to develop adverse effects or suffers a relapse when another preparation is substituted.
Route of Administration. Different routes of administration can affect the rate of absorption as well as the ratio of parent compound to its various metabolites. For example, the concentration versus time curve is usually shifted to the left with the intramuscular route due to more rapid absorption (Fig. 2-1). Hence, Tmax is shifted to the left (i.e., shortened) and Cmax is higher, contracting the curve, even though the AUC may be unchanged.
Route of Administration. Different routes of administration can affect the rate of absorption as well as the ratio of parent compound to its various metabolites. For example, the concentration versus time curve is usually shifted to the left with the intramuscular route due to more rapid absorption (Fig. 2-1). Hence, Tmax is shifted to the left (i.e., shortened) and Cmax is higher, contracting the curve, even though the AUC may be unchanged.
This pattern is not true for all drugs. For example, diazepam and chlordiazepoxide are unstable at a pH of 7.4, tend to crystallize in tissue when given intramuscularly, and are less bioavailable through this route versus oral administration (13,16,17). Their absorption also tends to be erratic and variable, depending on where the injection was given (i.e., near blood vessels, in fat, or in muscle), as well as slower and less complete.
First-Pass Effect. When drugs are administered orally, they typically are absorbed in the small bowel, enter the portal circulation, and then pass through the liver. Both CYP isoenzymes in the bowel wall and in the liver can metabolize a portion of the drug before it reaches the systematic circulation (i.e., first-pass metabolism or first-pass effect). The extent of this effect can be broadly altered by diseases (e.g., cirrhosis, portacaval shunting, persistent hepatitis, and congestive heart failure) and by some drugs (e.g., alcohol and ketoconazole), influencing the peak concentrations achieved and the ratio of the parent compound to metabolites (18,19).
Transporters can either actively take up or extrude drugs, increasing or decreasing absorption, respectively. For example, P-gp (an ATPdependent efflux pump with broad substrate specificity) transports various compounds across the cell membrane. This serves to regulate the distribution and bioavailability of certain drugs; to remove toxic metabolites and xenobiotics from cells into urine, bile, and intestinal lumen; and to transfer compounds out of the brain across the BBB. Administered drugs or dietary factors can increase or decrease the activity of transporters, altering the degree of absorption of another drug.
Once first-pass metabolism occurs, metabolites are excreted into the bile and then the small bowel. Those that are lipid-soluble are reabsorbed into the portal circulation, eventually entering the systemic circulation. These metabolites may have a similar or substantially different pharmacological profile from their parent drug. For example, chlorpromazine undergoes extensive hepatic biotransformation and has 168 theoretical metabolites, 70 of which have been identified in plasma and tissue. Some have dopamine receptor-blocking activity, although they are weaker than the parent compound, making it difficult to separate its effects (good or bad) from its numerous active metabolites.
In contrast to oral administration, drugs administered intravenously or intramuscularly enter the systemic circulation directly, avoiding the first-pass effect. This is why some drugs (e.g., acute parenteral olanzapine, ziprasidone, or aripiprazole; risperidone microspheres) are more potent (i.e., greater effect per milligram dose taken) when administered intramuscularly and their dose adjusted to compensate for this difference in absorption. Medical conditions such as cirrhosis can cause portacaval shunting, allowing drugs to avoid the first-pass effect and enter the systemic circulation directly, enhancing their effect. Coadministered drugs can affect first-pass metabolism or transport of another drug, altering its bioavailability (7,14,20). For example, acute alcohol intoxication can substantially reduce the first-pass effect on TCAs, leading to a doubling of the Cmax after the same dose (21). This effect contributes to the increased toxicity that occurs when an overdose of TCAs is taken with alcohol.
Distribution
Once in the systemic circulation, drugs distribute to organs in direct proportion to their fat and protein content (22). The rate of accumulation is a function of an organ’s vascularity. Although highly lipid-soluble drugs accumulate in adipose tissue to the same extent that they accumulate in the brain, the rate of accumulation is much faster in the brain than in adipose tissue.
Figure 2-2 illustrates single and multiple dose pharmacokinetics, with the Y axis representing plasma drug concentration and the × axis the time elapsed since drug administration. When a single oral dose is administered, the drug reaches Cmax and then undergoes a relatively rapid decline. This initial decline is due primarily to drug distribution, rather than to elimination.
Thus, the initial drug concentration drop is a function of the rate of uptake into other bodily compartments rather than elimination from the body. With the first dose of an intravenously administered psychotropic, this is particularly important because psychotropics
Thus, the initial drug concentration drop is a function of the rate of uptake into other bodily compartments rather than elimination from the body. With the first dose of an intravenously administered psychotropic, this is particularly important because psychotropics
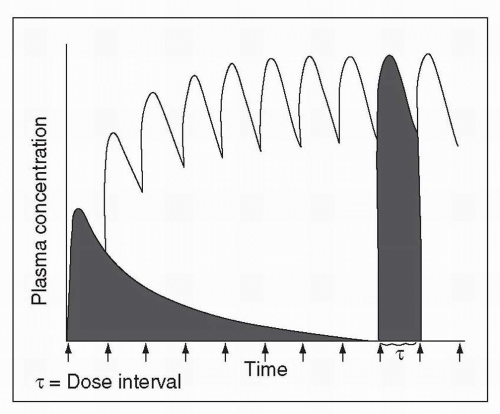 Figure 2-2 Plasma drug concentration versus time curve following administration of a single oral dose and following repeated administrations of the same oral dose. |
Are quite lipophilic
Have large volumes of distribution
Reach tissue concentrations typically 10 to 100 times greater than their plasma concentrations
The acute effects of a single dose of most psychotropics are terminated by redistribution. An example would be the acute sedative effects of intravenously administered lorazepam, which rapidly enters the brain from the blood. A disproportionate amount of the dose enters the CNS because of its greater vascularity in comparison with peripheral adipose tissue. Brain concentration subsequently falls, as the drug redistributes into the plasma and then into other peripheral compartments. This fall in brain concentration terminates the acute psychoactive effects of lorazepam.
Case Example
A 44-year-old chronic alcoholic man presented in the emergency room with disorientation and combativeness after 2 days of abstinence. He complained of visual and tactile hallucinations and was found to have elevated heart rate, blood pressure, and temperature. Lorazepam was slowly administered intravenously, and after 15 minutes, the patient was mildly sedated. He was then transferred to an inpatient unit. During this 30-minute interval, he received no additional lorazepam, and when he arrived on the floor, his symptoms had returned. He became agitated, struck one of the nursing staff, and had to be physically restrained. In this case, failure to account for the phenomenon of drug redistribution resulted in a potentially unavoidable adverse outcome.
Distribution of the drug is conceptualized as accumulation into various body compartments (e.g., fat, aqueous, bone, and brain). The extent to which drugs differ in their rate and degree of accumulation is related to the number of compartments into which they equilibrate. Even within one apparent compartment, such as blood, there may be more than one subcomponent for distribution, including
Plasma water
Plasma protein
Circulating cells (particularly red blood cells)
Most psychotropics are highly protein bound (23,24,25 and 26) and these bound agents often account for more than 90% of the total plasma concentration. The clinical significance is that even though the free-drug fraction is the smallest absolute amount, it is the most important because its concentration determines the final equilibration with the site of action. Although a change in the bound amount from 95% to 90% may seem small, the corresponding change in the free fraction would be from 5% to 10%, doubling the effective drug concentration in equilibrium with the site of action. Thus, any condition that shifts the ratio of bound to free drug can change the concentration at the sites of action and thus the magnitude of effect. Conditions that lead to a functional decrease in the amount of circulating protein include
Malnutrition, as with severe anorexia
Wasting, as in the nephrotic syndrome
Increasing the relative amount of the free fraction can increase toxicity. Most assays used in routine TDM (see “Therapeutic Drug Monitoring,” later in this chapter) do not distinguish between bound and free-drug fractions and will not detect such changes unless specialized techniques are used.
Acute and chronic inflammatory processes can increase the amount of circulating α1-acid glycoprotein (which avidly binds a variety of psychotropics), increasing the absolute amount of bound drug, while the free fraction remains unchanged. In such circumstances, the circulating total concentrations may seem excessive but actually simply represent an increase in the bound (and biologically inert) circulating fraction (30).
The absolute and the relative size of the body’s fat compartment can change with normal aging and morbid obesity (19,22,31,32 and 33). As noted earlier, the percentage of total body water and protein content decreases in the elderly, whereas the percentage of fat content increases, providing a relatively larger reservoir in which to store psychotropics. These changes explain why many agents have a more persistent effect in the elderly. The morbidly obese patient also has an increased reservoir, and a drug’s effect can persist in relation to the size of the adipose compartment.
Case Exampl
A 62-year-old man presented to his internist with a major depressive disorder. His past medical history was significant for two previous myocardial infarctions and related congestive heart failure, well controlled by digoxin. There was also a past history of significant alcohol abuse, in remission for the past 4 years. The internist prescribed amitriptyline (75 mg by mouth at bedtime). One week later, the patient returned with a worsening of his depressive state, and the internist increased the dose to 100 mg. The patient continued to deteriorate and was referred to a psychiatrist, who hospitalized him. The patient was now suspicious, guarded, and irritable, receiving a diagnosis of a psychotic depression. Haloperidol (10 mg by mouth at bedtime) was added. Five days later, the patient was found unconscious in his room after complaining of faintness and heart palpitations. He was rushed to the coronary intensive care unit, where a 2:1 atrioventricular block and periodic runs of premature ventricular contractions were found on the electrocardiogram (ECG). Drug level monitoring revealed a total TCA plasma level of 950 ng/mL. All psychoactive drugs were stopped and as the plasma level fell, the cardiac and the psychotic symptoms resolved. This patient had several risk factors for the development of toxic TCA concentrations even while on a relatively low dose of amitriptyline.
Under steady-state conditions, there is a proportional relationship between the tissue and the plasma compartments (4,15,34). This underlies the usefulness of TDM, which uses drug concentration in plasma to ensure adequacy of dose while helping to avoid toxicity. Although psychotropics do not exert their effects in plasma, their plasma concentration is, nonetheless, in equilibrium with tissue levels. Although tissue concentrations (depending on the organ) are 10 to 100 times greater than plasma concentrations, the latter provide an indirect measurement of the former (2,34).
Metabolism
Biotransformation. Most psychotropics undergo extensive oxidative biotransformation leading to the formation of more polar metabolites, which are then excreted in the urine. Biotransformation may involve one or more of the following steps:
Hydroxylation
Demethylation
Oxidation
Sulfoxide formation
Although most drugs undergo extensive oxidative biotransformation (i.e., phase I metabolism) before elimination, some undergo simple conjugation with moieties such as glucuronic acid (i.e., phase II metabolism), and others are excreted unmetabolized (e.g., lithium) (35). Because conjugation can occur in most organs, phase II metabolism is not dependent on liver function. Thus, the clearance of drugs that undergo only glucuronidation is generally not affected by significant liver impairment. These include all of the 3-hydroxybenzodiazepines (e.g., lorazepam, oxazepam, and temazepam), which are as readily cleared by the old as by the young, as long as renal function is normal. The same is true for patients with severely compromised liver function, which is another reason to use 3-hydroxybenzodiazepines (e.g., lorazepam) early in the treatment of delirium tremens. Should such patients subsequently develop hepatic failure, they can still readily clear such drugs and avoid having them persist in the body to worsen cognitive function and level of arousal. By contrast, diazepam requires extensive biotransformation as a necessary step in its elimination and can persist in the body for a prolonged period should the patient develop hepatic failure (17). Further, the metabolites of diazepam have similar pharmacological properties, accumulate in the body, and ultimately contribute to its pharmacological effect.
Oxidative biotransformation results in the formation of metabolites whose pharmacological effects may be similar or dissimilar to the parent compound. Either way, active metabolites contribute to the final overall clinical effects. For example, norfluoxetine has essentially the same activity as fluoxetine in terms of both serotonin uptake blockade and inhibition of several CYP isoenzymes but is cleared more slowly (35). As a result, norfluoxetine accumulates extensively in the body following chronic administration of fluoxetine, making it, rather than the parent compound, the principal determinant of the clinical effect.
In contrast, clomipramine’s major metabolite, desmethylclomipramine, has a markedly different pharmacological profile from the parent drug (36). Although clomipramine is a potent inhibitor of serotonin uptake, desmethylclomipramine is a more potent inhibitor of norepinephrine uptake. If clomipramine’s value in obsessive-compulsive disorder (OCD) depends on its ability to block 5-HT uptake (and not of norepinephrine), then this therapeutic effect should be a function of the relative ratio between clomipramine and desmethylclomipramine. Thus, clomipramine could lose its effectiveness for OCD if a patient was a more efficient demethylator of the parent drug.
Another important situation occurs when the parent compound is biotransformed into a less efficacious and possibly more toxic metabolite. For example, if the concentration of the hydroxylated metabolite of imipramine (2-hydroxyimipramine) is increased, this TCA could lose its effectiveness while simultaneously increasing in toxicity (37).
Cytochrome P450 Enzyme Induction. Alcohol, nicotine, and most anticonvulsants induce one or more CYP isoenzymes (38,39). This mechanism explains why barbiturates and carbamazepine (CBZ) induce the metabolism of other drugs as well as their own (i.e., autoinduction), since they are substrates of the same CYP enzyme they induce. Blood levels obtained 3 or 4 days after starting these drugs reflect the rate of elimination at that time; however, levels will subsequently fall on the same dose because autoinduction results in faster elimination (i.e., a shorter half-life) as a function of continued drug administration. Therefore, early TDM of CBZ will overestimate the eventual concentration reached after several weeks on this drug (see Chapter 10).
Alcohol has a triphasic effect on the elimination rates of drugs that require extensive biotransformation (21). For example, acute alcohol ingestion can significantly block the metabolism of high first-pass clearance drugs (e.g., TCAs), substantially increasing their bioavailability and their adverse effects (e.g., increased lethality of a TCA overdose when taken together with alcohol).
Drinking alcohol on a regular basis for several weeks to months can induce CYP isoenzymes such as 1A2 and 3A3/4, resulting in a lower plasma concentration of drugs dependent on these enzymes for their clearance (e.g., olanzapine, a CYP 1A2 substrate; quetiapine, a CYP 3A3/4 substrate). Thus, subacute and subchronic alcohol ingestion induces liver enzymes and causes lower plasma levels of drugs that undergo oxidative biotransformation as a necessary step in their elimination.
Chronic alcohol ingestion may cause cirrhosis, reducing hepatic CYP enzyme concentration and liver mass and causing portacaval shunting. These effects will result in increased plasma drug levels due to both greater bioavailability (resulting from reduced first-pass metabolism) and decreased clearance. Thus, dose adjustment is necessary and should be guided by TDM when possible.
Cytochrome P450 Enzyme Inhibition. In contrast to anticonvulsants and alcohol, drugs such as fluoxetine, paroxetine, fluvoxamine, bupropion, nefazodone, quinidine, and some antipsychotics can inhibit specific CYP isoenzymes (7,35,36,40,41). Thus, drugs whose metabolism is dependent on these enzymes (e.g., many of the antipsychotics, antidepressants, and anxiolytics discussed later) can have their clearance reduced by the coadministration of agents such as fluoxetine. For example, fluoxetine at 20 mg/day produces on average a 500% increase in the levels of coprescribed drugs that are principally dependent on CYP 2D6 for their clearance (42). That can lead to an increased incidence and severity of dose- and concentration-dependent adverse effects. Such a situation may even be life threatening if the drug has a narrow therapeutic index and the dose is not adjusted to compensate for the change in its clearance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


