KEY POINTS
In infants with Bochdalek-type congenital diaphragmatic hernia, the severity of pulmonary hypoplasia and the resultant pulmonary hypertension are key determinants of survival. Barotrauma and hypoxia should be avoided.
During initial management of an infant with esophageal atresia and distal tracheoesophageal fistula, every effort should be made to avoid distending the gastrointestinal tract, especially when using mechanical ventilation. The patient should be evaluated for components of the VACTERRL (vertebral, anorectal, cardiac, tracheoesophageal, renal, radial limb) anomalies. Timing and extent of surgery are dictated by the stability of the patient.
Although malrotation with midgut volvulus occurs most commonly within the first few weeks of life, it should always be considered in the differential diagnosis in a child with bilious emesis. Volvulus is a surgical emergency; therefore, in a critically ill child, prompt surgical intervention should not be delayed for any reason.
When evaluating a newborn infant for vomiting, it is critical to distinguish between proximal and distal causes of intestinal obstruction using both prenatal and postnatal history, physical examination, and abdominal radiographs.
Risk factors for necrotizing enterocolitis (NEC) include prematurity, formula feeding, bacterial infection, and intestinal ischemia. Critical to the management of infants with advanced (Bell stage III) or perforated NEC is timely and adequate source control of peritoneal contamination. Early sequelae of NEC include perforation, sepsis, and death. Later sequelae include short bowel syndrome and stricture.
In patients with intestinal obstruction secondary to Hirschsprung’s disease, a leveling ostomy or endorectal pull-through should be performed using ganglionated bowel, proximal to the transition zone between ganglionic and aganglionic intestine.
Prognosis of infants with biliary atresia is directly related to age at diagnosis and timing of portoenterostomy. Infants with advanced age at the time of diagnosis or infants who fail to demonstrate evidence of bile drainage after portoenterostomy usually require liver transplantation.
Infants with omphaloceles have greater associated morbidity and mortality than infants with gastroschisis due to a higher incidence of congenital anomalies and pulmonary hypoplasia. Gastroschisis can be associated with intestinal atresia, but not with other congenital anomalies. An intact omphalocele can be repaired electively, whereas gastroschisis requires urgent intervention to protect the exposed intestine.
Prognosis for children with Wilms’ tumor is defined by the stage of disease at the time of diagnosis and the histologic type (favorable vs. unfavorable). Preoperative chemotherapy is indicated for bilateral involvement, a solitary kidney, or tumor in the inferior vena cava above the hepatic veins. Gross tumor rupture during surgery automatically changes the stage to 3 (at a minimum).
Injury is the leading cause of death in children older than 1 year of age. Blunt mechanisms account for the majority of pediatric injuries. The central nervous system is the most commonly injured organ system and the leading cause of death in injured children.
INTRODUCTION
In his 1953 classic textbook entitled The Surgery of Infancy and Childhood, Dr. Robert E. Gross summarized the essential challenge of pediatric surgery: “Those who daily operate upon adults, even with the greatest of skill, are sometimes appalled – or certainly are not at their best – when called upon to operate upon and care for a tiny patient. Something more than diminutive instruments or scaled-down operative manipulations are necessary to do the job in a suitable manner.” To this day, surgical residents and other trainees often approach the pediatric surgical patient with the same mix of fear, trepidation, and anxiety. Importantly, these same trainees typically complete their pediatric surgical rotations with a profound respect for the resilience of young children to undergo complex operations, and with an appreciation for the precision required from their caregivers, both in the operating room and during the perioperative period. Over the decades, the specialty of pediatric surgery has evolved considerably to encompass not only the care of the smallest patients with surgical disorders, who may require in utero interventions in certain circumstances, but also the care of older infants, children, adolescents, and young adults. Similarly, our understanding of the pathophysiology of the diseases that pediatric surgeons face has evolved to the point that some pediatric surgical diseases are now understood at the level of molecular or cellular signaling pathways. Pediatric surgery provides the opportunity to intervene positively in a wide array of diseases and to exert a long-lasting impact on the lives of children and their grateful parents. The scope of diseases encountered in the standard practice of pediatric surgery is immense and includes anomalies of the head and neck, thoracic, gastrointestinal, and genitourinary areas. This chapter is not designed to cover the entire spectrum of diseases a pediatric surgeon is expected to master; rather, it presents a synopsis of a handful of pediatric surgical conditions that a practicing general surgeon is likely to encounter over the course of his or her career.
PEDIATRIC SURGICAL THEMES: PITFALLS AND PEARLS
This chapter focuses on the unique considerations regarding the diagnosis and management of surgical diseases in the pediatric population. Many surgical trainees approach the surgical care of children with some degree of fear and trepidation. As any pediatric caregiver will attest to, the surgical management of infants and children requires delicate, careful, and professional interactions with their parents. The stress that the parents of sick children experience in the hospital setting can, at times, be overwhelming. It is due, in part, to the uncertainty regarding a particular prognosis, the feeling of helplessness that evolves when one is unable to care for one’s own child, and in certain cases, the guilt or remorse that one feels for not seeking medical care earlier or for consenting to a particular procedure. Management of the sick child and his or her family requires not only a certain set of skills, but also a unique knowledge base. This section is included to summarize some important general principles in accomplishing this task.
Children are not little adults, but they are little people. In practical terms, this often-heard refrain implies that children have unique fluid, electrolyte, and medication needs. Thus, the dosage of medications and the administration of intravenous fluids should be based on their weight. The corollary of this point is that infants and young children are extremely sensitive to perturbations in their normal physiology, and as such may easily experience fluid overload or dehydration.
Sick children whisper before they shout. Children with surgical diseases can deteriorate very quickly. But before they deteriorate, they often manifest subtle physical findings. These findings—referred to as “whispers”—may include signs such as tachycardia, bradycardia, hypothermia, fever, recurrent emesis, or feeding intolerance. Meticulous attention to these subtle findings may unmask the development of potentially serious, life-threatening physiologic disturbances.
Always listen to the mother and the father. Surgical diseases in children can be very difficult to diagnose because children are often minimally communicative and information that they communicate may be confusing, conflicting, or both. In all cases, it is wise to listen to the child’s parents, who have closely observed their child and know him or her best. Most importantly, the child’s parents know with certainty whether or not the child is sick despite not always knowing the precise diagnosis.
Children suffer pain after surgery. Timely and adequate pain management must accompany surgical interventions.
Pediatric tissue must be handled delicately and with profound respect.
GENERAL CONSIDERATIONS
In managing the pediatric surgical patient, an understanding of fluid and electrolyte balance is critical, as the margin between dehydration and fluid overload is rather small. This is particularly true in infants, who have little reserve when ill. Failure to pay meticulous attention to their hydration status can result in significant fluid overload or dehydration. Several surgical diagnoses such as gastroschisis or short-gut syndrome are characterized by a predisposition to fluid loss. Others require judicious restoration of intravascular volume in order to prevent cardiac failure, as is the case in patients with congenital diaphragmatic hernia and associated pulmonary hypertension. The infant’s physiologic day is approximately 8 hours in duration. Accordingly, careful assessment of the individual patient’s fluid balance, including fluid intake and output for the previous 8 hours, is essential to prevent dehydration or fluid overload. Clinical signs of dehydration include tachycardia, decreased urine output, reduced skin turgor, a depressed fontanelle, absent tears, lethargy, and poor feeding. Fluid overload is often manifested by the onset of new oxygen requirement, respiratory distress, tachypnea, and tachycardia. The physical assessment of the fluid status of each child must include a complete head-to-toe evaluation, with emphasis on determining whether perturbations in normal physiology are present.
At 12 weeks’ gestation, the total body water of a fetus is approximately 94 cc/kg. By the time the fetus reaches full term, the total body water has decreased to approximately 80 cc/kg. Total body water drops an additional 5% within the first week of life, and by 1 year of life, total body water approaches adult levels, around 60 to 65 cc/kg. Parallel to the drop in total body water is the reduction in extracellular fluid. These changes are accelerated in the preterm infant who may face additional fluid losses due to coexisting congenital anomalies or surgery. Normal daily maintenance fluids for most children can be estimated using the following formula: 100 mL/kg for the first 10 kg, plus 50 mL/kg for 11 to 20 kg, plus 25 mL/kg for each additional kilogram of body weight thereafter. Because intravenous (IV) fluid orders are written as milliliters per hour, this can be conveniently converted as follows: 4 mL/kg/h up to 10 kg, add 2 mL/kg/h for 11 to 20 kg, and add 1 mL/kg/h for each additional kilogram of body weight thereafter. For example, a 26-kg child has an estimated maintenance fluid requirement of (10 × 4) + (10 × 2) + (6 × 1) = 66 mL/h in the absence of massive fluid losses or shock. A newborn infant with gastroschisis will manifest significant evaporative losses from the exposed bowel such that fluid requirements may range from 150 to 180 cc/kg/d. Precise management of a neonate’s fluid status requires an understanding of changes in the glomerular filtration rate (GFR) and tubular function of the kidney. The term newborn’s GFR is approximately 21 mL/min/1.73 m2 compared to 70 mL/min/1.73 m2 in an adult. Within the first 2 weeks of life, GFR increases to approximately 60, and by 2 years of age, it is essentially at adult levels. The capacity to concentrate urine is very limited in preterm and term infants. Compared with an adult who can concentrate urine to 1200 mOsm/kg, infants can concentrate urine at best to 600 mOsm/kg. While infants are capable of secreting antidiuretic hormone (ADH), the aquaporin water channel–mediated osmotic water permeability of the infant’s collecting tubules is severely limited compared with that of adults, leading to insensitivity to ADH.
Sodium requirements range from 2 mEq/kg/d in term infants up to 5 mEq/kg/d in critically ill preterm infants as a consequence of salt wasting. Potassium requirements are on the order of 1 to 2 mEq/kg/d. Calcium and magnesium supplementation of IV fluids is essential to prevent laryngospasm, dysrhythmias, and tetany.
Acute metabolic acidosis usually implies inadequate tissue perfusion and is a serious disorder in children. Potentially life-threatening causes that are specific for the pediatric population must be sought; they include intestinal ischemia from necrotizing enterocolitis (in the neonate), midgut volvulus, or incarcerated hernia. Other causes include chronic bicarbonate loss from the gastrointestinal tract or acid accumulation as in chronic renal failure. Respiratory acidosis implies hypoventilation, the cause of which should be apparent. Treatment of acute metabolic acidosis should be aimed at restoring tissue perfusion by addressing the underlying abnormality first. For severe metabolic acidemia where the serum pH is less than 7.25, sodium bicarbonate should be administered using the following guideline: base deficit × weight in kilograms × 0.5 (in newborns). The last factor in the equation should be 0.4 for smaller children and 0.3 for older children. The dose should be diluted to a concentration of 0.5 mEq/mL because full-strength sodium bicarbonate is hyperosmolar. One half of the corrective dose is given, and the serum pH is measured again. During cardiopulmonary resuscitation (CPR), one half of the corrective dose can be given as an IV bolus and the other half given slowly via IV.
Respiratory alkalosis is usually caused by hyperventilation, which is readily correctable. Metabolic alkalosis most commonly implies gastric acid loss, as in the child with pyloric stenosis, or aggressive diuretic therapy. In the child with gastric fluid loss, IV fluids of 5% dextrose, 0.5% normal saline, and 20 mEq KCl/L usually correct the alkalosis.
Criteria for blood transfusion in infants and children remain poorly defined. The decision to transfuse a critically ill pediatric patient may depend on a number of clinical features that include the patient’s age, primary diagnosis, the presence of ongoing bleeding, coagulopathy, hypoxia, hemodynamic compromise, lactic acidosis, cyanotic heart disease, and overall severity of illness. A recent survey of transfusion practices among pediatric intensivists showed that the baseline hemoglobin levels that would prompt them to recommend packed red blood cell (PRBC) transfusion ranged from 7 to 13 g/dL. Patients with cyanotic heart disease are often transfused to higher hemoglobin values, although the threshold for transfusion in this population remains to be defined. To decrease the need for transfusion, other strategies have been considered. Studies in both critically ill adults and neonates have shown that administration of erythropoietin decreases PRBC transfusion requirements. In general terms, there is a trend toward an avoidance of the use of PRBC products whenever possible, as current studies suggest that lower hemoglobin concentrations are well tolerated by many groups of patients, whereas administration of PRBCs may have unintended negative consequences. In addition, there is increasing evidence that PRBC transfusion may have adverse effects on the host immune system, in both children and adults. These effects are poorly understood but may be due to red blood cell (RBC) storage or factors that are particular to the individual RBC donor.
A useful guideline for estimating blood volume for the newborn infant is approximately 80 mL/kg of body weight. When PRBCs are required, the transfusion requirement is usually administered in 10-mL/kg increments, which is roughly equivalent to a 500-mL transfusion for a 70-kg adult. The following formula may be used to determine the volume (mL) of PRBCs to be transfused:
In the child, coagulation deficiencies rapidly may assume clinical significance after extensive blood transfusion. It is advisable to have fresh frozen plasma and platelets available if more than 30 mL/kg have been transfused. Plasma is given in a dose of 10 to 20 mL/kg, and platelets are given in a dose of 1 unit/5 kg. Each unit of platelets consists of 40 to 60 mL of fluid (plasma plus platelets). Following transfusion of PRBCs to neonates with tenuous fluid balance, a single dose of a diuretic (such as furosemide 1 mg/kg) may help to facilitate excretion of the extra fluid load. Many clinicians prefer to administer fresh products to minimize the deleterious effects of red cell storage.
The nutritional requirements of the surgical neonate must be met in order for the child to grow and to heal surgical wounds. If inadequate protein and carbohydrate calories are given, the child may not only fail to recover from surgery, but may also exhibit growth failure and impaired development of the central nervous system. In general terms, the adequacy of growth must be assessed frequently, by determining both total body weight and head circumference. Neonates who are particularly predisposed to protein-calorie malnutrition include those with gastroschisis, intestinal atresia, or intestinal insufficiency from other causes, such as necrotizing enterocolitis. The protein and caloric requirements for the surgical neonate are shown in Table 39-1.
Nutrition can be provided via either the enteral or parenteral route. Whenever possible, the enteral route is preferred because it not only promotes the growth and function of the gastrointestinal system, but also ensures that the infant learns how to feed. There are various enteral feeding preparations available; these are outlined in Table 39-2. The choice of formula is based on the individual clinical state of the child. Pediatric surgeons are often faced with situations where oral feeding is not possible. This problem can be seen in the extremely premature infant who has not yet developed feeding skills or in the infant with concomitant craniofacial anomalies that impair sucking, for example. In these instances, enteral feeds can be administered either via a nasojejunal or a gastrostomy tube.
| FORMULA | kcal/mL | PROTEIN (g/mL) | FAT (g/mL) | CARBOHYDRATE (g/mL) |
|---|---|---|---|---|
| Human milk | 0.67 | 0.011 | 0.04 | 0.07 |
| Milk-based formula | ||||
| Enfamil 20 | 0.67 | 0.015 | 0.038 | 0.069 |
| Similac 20 | 0.67 | 0.015 | 0.036 | 0.072 |
| Soy-based formula | ||||
| Prosobee | 0.67 | 0.02 | 0.036 | 0.07 |
| Isomil | 0.67 | 0.018 | 0.037 | 0.068 |
| Special formula | ||||
| Pregestimil | 0.67 | 0.019 | 0.028 | 0.091 |
| Alimentum | 0.67 | 0.019 | 0.038 | 0.068 |
| Preterm | ||||
| Enfamil Premature | 0.80 | 0.024 | 0.041 | 0.089 |
When the gastrointestinal tract cannot be used because of mechanical, ischemic, inflammatory, or functional disorders, parenteral alimentation must be given. Prolonged parenteral nutrition is delivered via a central venous catheter. Peripheral IV alimentation can be given, using less concentrated but greater volumes of solutions. Long-term parenteral nutrition should include supplemental copper, zinc, and iron to prevent the development of trace metal deficiencies. A major complication of long-term total parenteral nutrition (TPN) is the development of parenteral nutrition-associated cholestasis, which can eventually progress to liver failure. To prevent this major complication, concomitant enteral feedings should be instituted and the gastrointestinal tract should be used as soon as possible. When proximal stomas are in place, gastrointestinal continuity should be restored as soon as possible. Where intestinal insufficiency is associated with dilation of the small intestine, tapering or intestinal lengthening procedures may be beneficial. Other strategies to minimize the development of TPN-related liver disease include meticulous catheter care to avoid infection, which increases cholestatic symptoms, aggressive treatment of any infection, and early cycling of parenteral nutrition in older children who can tolerate not receiving continuous dextrose solution for a limited period. Preliminary evidence suggests the possibility that substituting omega-3 fish oil lipid emulsion in parenteral nutrition for the standard soybean-based emulsions may prevent the development of TPN-related cholestasis and reverse the effects of established liver disease.
Obtaining reliable vascular access in an infant or child is an important task that often becomes the responsibility of the pediatric surgeon. The goal should always be to place the catheter in the least invasive, least risky, and least painful manner, and in a location that is most accessible and allows for use of the catheter without complications for as long as it is needed. In infants, central venous access may be established using a cutdown approach, either in the antecubital fossa, external jugular vein, facial vein, or proximal saphenous vein. If the internal jugular vein is used, care is taken to prevent venous occlusion. In infants over 3 kg and in older children, percutaneous access of the subclavian, internal jugular, or femoral veins is possible in most cases, and central access is achieved using the Seldinger technique. The catheters are tunneled to an exit site separate from the venotomy site. Where available, peripherally inserted central catheter (PICC) lines may be placed, typically via the antecubital fossa. Regardless of whether the catheter is placed by a cutdown approach or percutaneously, a chest x-ray to confirm central location of the catheter tip and to exclude the presence of a pneumothorax or hemothorax is mandatory. When discussing the placement of central venous catheters with parents, it is important to note that the complication rate for central venous lines in children can be high. The incidence of catheter-related sepsis or infection approaches 10% in many series, although recent reports show a lower incidence. Superior or inferior vena caval occlusion is a significant risk after the placement of multiple lines, particularly in the smallest premature patients.
Careful regulation of the ambient environment of infants and children is crucial, as these patients are extremely thermolabile. Premature infants are particularly susceptible to changes in environmental temperature. Because they are unable to shiver and lack stores of fat, their potential for thermogenesis is impaired. The innate inability to regulate temperature is compounded by the administration of anesthetic and paralyzing agents. Since these patients lack adaptive mechanisms to cope with the environment, the environmental temperature must be regulated. Attention to heat conservation during transport of the infant to and from the operating room is essential. Transport systems incorporating heating units are necessary for premature infants. In the operating room, the infant is kept warm by the use of overhead heating lamps, a heating blanket, warming the room as well as inspired gases, and coverage of the extremities and head with occlusive materials. During abdominal surgery, extreme care is taken to avoid wet and cold drapes. All fluids used to irrigate the chest or abdomen must be warmed to body temperature. Laparoscopic approaches for abdominal operations may result in more stable thermoregulation due to decreased heat loss from the smaller wound size. Constant monitoring of the child’s temperature is critical in a lengthy procedure, and the surgeon should continuously communicate with the anesthesiologist regarding the temperature of the patient. The development of hypothermia in infants and children can result in cardiac arrhythmias or coagulopathy. These potentially life-threatening complications can be avoided by careful attention to thermoregulation.
All children, including neonates, experience pain. Therefore, careful recognition and management of pediatric pain represents an important component of the perioperative management of all pediatric surgical patients. There is a range of pain management options that can improve the child’s well-being, as well as the parents’ sense of comfort. The use of a pacifier, which may be dipped in sucrose, has been shown to decrease crying time and neonatal pain scores after minor procedures. For situations where more pain is expected, IV narcotic agents should be used. Morphine and fentanyl have an acceptable safety margin and can be administered judiciously to neonates and children. A randomized clinical trial involving neonates on ventilators showed that the use of a morphine infusion decreased the incidence of intraventricular hemorrhage by 50%. Additional analgesic modalities include the use of topical anesthetic ointment (EMLA cream), regional anesthesia such as caudal blocks for hernias, and epidural or incisional catheter infusions (On-Q™) for large abdominal or thoracic incisions. In surgical neonates who have received large concentrations of narcotics over a prolonged period, transient physical dependence should not only be expected, but anticipated. When narcotics are discontinued, symptoms of narcotic withdrawal may develop, including irritability, restlessness, and episodes of hypertension and tachycardia. Early recognition of these signs is essential, as is timely treatment using naloxone and other agents. In the postoperative period, patient-controlled analgesia is another excellent method of pain control. By ensuring that the pediatric surgical patient has adequate analgesia, the surgeon ensures that the patient receives the most humane and thorough treatment and provides important reassurance to all other members of the healthcare team and to the family that pain control is a very high priority.
NECK MASSES
The management of neck masses in children is determined by their location and the length of time that they have been present. Neck lesions are found either in the midline or lateral compartments. Midline masses include thyroglossal duct remnants, thyroid masses, thymic cysts, or dermoid cysts. Lateral lesions include branchial cleft remnants, cystic hygromas, vascular malformations, salivary gland tumors, torticollis, and lipoblastoma (a rare benign mesenchymal tumor of embryonal fat occurring in infants and young children). Enlarged lymph nodes and rare malignancies such as rhabdomyosarcoma can occur either in the midline or laterally.
The most common cause of a neck mass in a child is an enlarged lymph node, which typically can be found laterally or in the midline. The patient is usually referred to the pediatric surgeon for evaluation after the mass has been present for several weeks. A detailed history and physical examination often helps determine the likely etiology of the lymph node and the need for excisional biopsy. Enlarged tender lymph nodes are usually the result of a bacterial infection (Staphylococcus or Streptococcus). Treatment of the primary cause (e.g., otitis media or pharyngitis) with antibiotics often is all that is necessary. However, when the involved nodes become fluctuant, incision and drainage are indicated. In many North American institutions, there has been an increasing prevalence of methicillin-resistant Staphylococcus aureus infection of the skin and soft tissues, leading to increased staphylococcal lymphadenitis in children. More chronic forms of lymphadenitis, including infections with atypical mycobacteria, as well as cat-scratch fever, are diagnosed based on serologic findings or excisional biopsy. The lymphadenopathy associated with infectious mononucleosis can be diagnosed based on serology. When the neck nodes are firm, fixed, and others are also present in the axillae or groin, or the history suggests lymphoma, excisional biopsy is indicated. In these cases, it is essential to obtain a chest radiograph to look for the presence of a mediastinal mass. Significant mediastinal load portends cardiorespiratory collapse due to loss of venous return and compression of the tracheobronchial tree with general anesthesia. Accordingly, such biopsies should be done under local anesthesia.
The thyroid gland buds off the foregut diverticulum at the base of the tongue in the region of the future foramen cecum at 3 weeks of embryonic life. As the fetal neck develops, the thyroid tissue becomes more anterior and caudad until it rests in its normal position. The “descent” of the thyroid is intimately connected with the development of the hyoid bone. Residual thyroid tissue left behind during the migration may persist and subsequently present in the midline of the neck as a thyroglossal duct cyst. The mass is most commonly appreciated in the 2- to 4-year-old child when the baby fat disappears and irregularities in the neck become more readily apparent. Usually the cyst is encountered in the midline at or below the level of the hyoid bone, and moves up and down with swallowing or with protrusion of the tongue. Occasionally it presents as an intrathyroidal mass. Most thyroglossal duct cysts are asymptomatic. If the duct retains its connection with the pharynx, infection may occur, and the resulting abscess will necessitate incision and drainage, occasionally resulting in a salivary fistula. Submental lymphadenopathy and midline dermoid cysts can be confused with a thyroglossal duct cyst. Rarely, midline ectopic thyroid tissue masquerades as a thyroglossal duct cyst and may represent the patient’s only thyroid tissue. Therefore, if there is any question regarding the diagnosis or if the thyroid gland cannot be palpated in its normal anatomic position, it is advisable to obtain a nuclear scan to confirm the presence of a normal thyroid gland. Although rarely the case in children, in adults the thyroglossal duct may contain thyroid tissue that can undergo malignant degeneration. The presence of malignancy in a thyroglossal cyst should be suspected when the cyst grows rapidly or when the ultrasound demonstrates a complex anechoic pattern or the presence of calcification.
If the cyst presents with an abscess, treatment should first consist of drainage and antibiotics. Following resolution of the inflammation, resection of the cyst in continuity with the central portion of the hyoid bone and the tract connecting to the pharynx in addition to ligation at the foramen cecum (the Sistrunk operation) is curative in over 90% of patients. Lesser operations result in unacceptably high recurrence rates, and recurrence is more frequent following infection. According to a recent review, factors predictive of recurrence included more than two infections prior to surgery, age under 2 years, and inadequate initial operation.
Paired branchial clefts and arches develop early in the fourth gestational week. The first cleft and the first, second, third, and fourth pouches give rise to adult organs. The embryologic communication between the pharynx and the external surface may persist as a fistula. A fistula is seen most commonly with the second branchial cleft, which normally disappears, and extends from the anterior border of the sternocleidomastoid muscle superiorly, inward through the bifurcation of the carotid artery, and enters the posterolateral pharynx just below the tonsillar fossa. In contrast, a third branchial cleft fistula passes posterior to the carotid bifurcation. The branchial cleft remnants may contain small pieces of cartilage and cysts, but internal fistulas are rare. A second branchial cleft sinus is suspected when clear fluid is noted draining from the external opening of the tract at the anterior border of the lower third of the sternomastoid muscle. Rarely, branchial cleft anomalies occur in association with biliary atresia and congenital cardiac anomalies, an association that is referred to as Goldenhar’s complex.
Complete excision of the cyst and sinus tract is necessary for cure. Dissection of the sinus tract is facilitated with passage of a fine lacrimal duct probe through the external opening into the tract and using it as a guide for dissection. Injection of a small amount of methylene blue dye into the tract also may be useful. A series of two or sometimes three small transverse incisions in a “stepladder” fashion is preferred to a long oblique incision in the neck, which is cosmetically undesirable. Branchial cleft cysts can present as abscesses. In these cases, initial treatment includes incision and drainage with a course of antibiotics to cover Staphylococcus and Streptococcus species, followed by excision of the cyst after the infection resolves.
Lymphatic malformation (previously known as cystic hygroma or lymphangioma) occurs as a result of sequestration or obstruction of developing lymph vessels in approximately 1 in 12,000 births. Although the lesion can occur anywhere, the most common sites are in the posterior triangle of the neck, axilla, groin, and mediastinum. The cysts are lined by endothelium and filled with lymph. Occasionally unilocular cysts occur, but more often, there are multiple cysts “infiltrating” the surrounding structures and distorting the local anatomy. A particularly troublesome variant of lymphatic malformation is one that involves the tongue, floor of the mouth, and structures deep in the neck. Adjacent connective tissue may show extensive lymphocytic infiltration. The mass may be apparent at birth or may appear and enlarge rapidly in the early weeks or months of life as lymph accumulates; most present by age 2 years (Fig. 39-1A). Extension of the lesion into the axilla or mediastinum occurs about 10% of the time and can be demonstrated preoperatively by chest x-ray, ultrasound (US), or computed tomography (CT) scan. Occasionally lymphatic malformations contain nests of vascular tissue. These poorly supported vessels may bleed and produce rapid enlargement and discoloration of the mass. Infection within the cysts, usually caused by Streptococcus or Staphylococcus, may occur. In the neck, this can cause rapid enlargement, which may result in airway compromise. Rarely, it may be necessary to carry out percutaneous aspiration of a cyst to relieve respiratory distress.
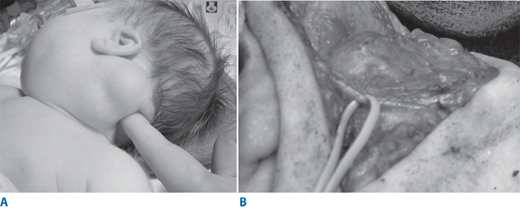
The diagnosis of lymphatic malformation by prenatal US, before 30 weeks’ gestation, has detected a “hidden mortality” as well as a high incidence of associated anomalies, including abnormal karyotypes and hydrops fetalis. Occasionally, very large lesions can cause obstruction of the fetal airway. Such obstruction can result in the development of polyhydramnios by impairing the ability of the fetus to swallow amniotic fluid. In these circumstances, the airway is usually markedly distorted, which can result in immediate airway obstruction unless the airway is secured at the time of delivery. Orotracheal intubation or emergency tracheostomy while the infant remains attached to the placenta, the so-called EXIT procedure (ex utero intrapartum treatment), may be necessary to secure the airway.
The modern management of most lymphatic malformations includes the combination of surgical excision and image-guided sclerotherapy. The initial treatment typically involves surgery in an attempt to safely remove all gross disease without damaging vital structures. Total removal of all gross disease may not be possible because of the extent of the lymphatic malformation and its proximity to, and intimate relationship with, adjacent nerves, muscles, and blood vessels (Fig. 39-1B). Radical ablative surgery is not indicated for this lesion. A combined sclerotherapy/resectional approach is particularly useful for large lymphatic malformations and those that extend to the base of the tongue or the floor of the mouth. Conservative excision and unroofing of the cysts is advised along with sclerotherapy or repeated partial excision for any residual lymphatic malformation if necessary, preserving all adjacent crucial structures. Postoperative wound drainage is important and is best accomplished by closed-suction technique. Nevertheless, fluid may accumulate beneath the surgically created flaps, requiring multiple needle aspirations.
The presence of a lateral neck mass in infancy in association with rotation of the head toward the opposite side of the mass indicates the presence of congenital torticollis. This lesion results from fibrosis of the sternocleidomastoid muscle. The mass may be palpated in the affected muscle in approximately two thirds of cases. Histologically, the lesion is characterized by the deposition of collagen and fibroblasts around atrophied muscle cells. In the vast majority of cases, physical therapy based on passive stretching of the affected muscle is of benefit. Rarely, surgical transection of the sternocleidomastoid may be indicated.
RESPIRATORY SYSTEM
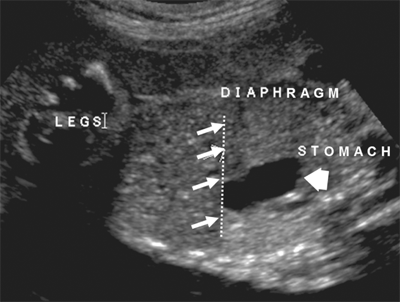
The septum transversum extends to divide the pleural and coelomic cavities during fetal development. This precursor of the diaphragm normally completes separation of these two cavities at the posterolateral aspects of this mesenchymally derived structure. The most common variant of a congenital diaphragmatic hernia is a posterolateral defect, also known as a Bochdalek hernia. Diaphragmatic defects allow abdominal viscera to fill the chest cavity. The abdominal cavity is small and underdeveloped and remains scaphoid after birth. Both lungs are hypoplastic, with decreased bronchial and pulmonary artery branching. Lung weight, lung volume, and DNA content are also decreased, and these findings are more striking on the ipsilateral side. This anomaly is encountered more commonly on the left (80%–90%). Linkage analyses have recently implicated genetic mutations in syndromic variants of congenital diaphragmatic hernias. In many instances, there is a surfactant deficiency, which compounds the degree of respiratory insufficiency. Amniocentesis with karyotype may identify chromosomal defects, especially trisomy 18 and 21. Associated anomalies, once thought to be uncommon, were identified in 65 of 166 patients in one study, predominantly of the heart, followed by abdominal wall defects, chromosomal changes, and other defects. Prenatal ultrasonography is successful in making the diagnosis of congenital diaphragmatic hernia (CDH) as early as 15 weeks’ gestation, and early antenatal diagnosis is associated with worse outcomes. US findings include herniated abdominal viscera in the chest, which may also look like a mass or lung anomaly, changes in liver position, and mediastinal shift away from the herniated viscera (Fig. 39-2). Accurate prenatal prediction of outcome for fetuses who have CDH is very difficult. One index of severity for patients with left CDH is the lung-to-head ratio (LHR), which is the product of the length and the width of the right lung at the level of the cardiac atria divided by the head circumference (all measurements in millimeters). An LHR value of less than 1.0 is associated with a very poor prognosis, whereas an LHR greater than 1.4 predicts a more favorable outcome. The utility of the LHR in predicting outcome in patients with CDH has recently been questioned, due to the tremendous interobserver variability in calculating this ratio for a particular patient, as well as the lack of reliable measures to determine postnatal disease severity.
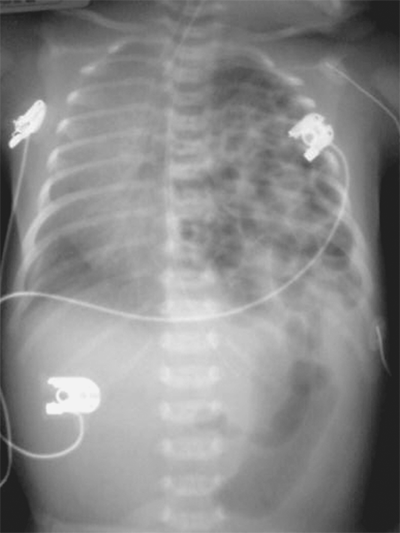
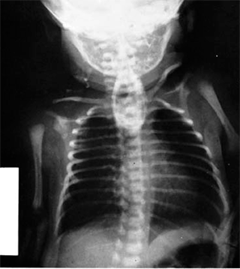
Following delivery, the diagnosis of CDH is made by chest x-ray (Fig. 39-3). The differential diagnosis includes bronchopulmonary foregut malformations, in which the intrathoracic loops of bowel may be confused for lung or foregut pathology. The vast majority of infants with CDH develop immediate respiratory distress, which is due to the combined effects of three factors. First, the air-filled bowel in the chest compresses the mobile mediastinum, which shifts to the opposite side of the chest, compromising air exchange in the contralateral lung. Second, pulmonary hypertension develops. This phenomenon results in persistent fetal circulation, with resultant decreased pulmonary perfusion and impaired gas exchange. Finally, the lung on the affected side is often hypoplastic, such that it is essentially nonfunctional. Varying degrees of pulmonary hypoplasia on the opposite side may compound these effects. The second and third factors are thought to be the most important. Neonates with CDH are usually in respiratory distress requiring ventilation and intensive care, and the overall mortality in most series is around 50%.
CDH care has improved considerably through effective use of improved methods of ventilation and timely cannulation for extracorporeal membrane oxygenation (ECMO). Many infants are symptomatic at birth due to hypoxia, hypercarbia, and metabolic acidosis. Prompt cardiorespiratory stabilization is mandatory. It is noteworthy that in some infants, the first 24 to 48 hours after birth are often characterized by a period of relative stability with high levels of PaO2 and relatively good perfusion. This has been termed the “honeymoon period” and is often followed by progressive cardiorespiratory deterioration. In the past, correction of the hernia was believed to be a surgical emergency, and patients underwent surgery shortly after birth. It is now accepted that the presence of persistent pulmonary hypertension that results in right-to-left shunting across the patent foramen ovale or the ductus arteriosus and the degree of pulmonary hypoplasia are the leading causes of cardiorespiratory insufficiency. Current management therefore is directed toward managing the pulmonary hypertension and minimizing barotrauma while optimizing oxygen delivery. To achieve this goal, infants are placed on mechanical ventilation using relatively low or “gentle” settings that prevent overinflation of the noninvolved lung. Levels of PaCO2 in the rangeof 50 to 60 mmHg or higher are accepted as long as the pH remains ≥ 7.25. If these objectives cannot be achieved using conventional ventilation, high-frequency oscillatory ventilation (HFOV) may be used to avoid the injurious effects of conventional tidal volume ventilation. Echocardiography will assess the degree of pulmonary hypertension and identify the presence of any coexisting cardiac anomaly. Intensive care unit goals include minimal sedation, meticulous attention to endotracheal tube secretions, and gradual changes to ventilator settings to avoid inducing pulmonary hypertension via hypoxia. To minimize the degree of pulmonary hypertension, inhaled nitric oxide may be administered and, in some patients, improves pulmonary perfusion. Nitric oxide is administered into the ventilation circuit and is used in concentrations up to 40 parts per million. Correction of acidosis using bicarbonate solution may minimize the degree of pulmonary hypertension. As the degree of pulmonary hypertension becomes hemodynamically significant, right-sided heart failure develops and systemic perfusion is impaired. Administration of excess IV fluid will compound the degree of cardiac failure and lead to marked peripheral edema. Inotropic support using epinephrine, dopamine, and milrinone alone or in combination may be useful in optimizing cardiac contractility and maintaining mean arterial pressure.
Infants with CDH who remain severely hypoxic despite maximal ventilatory care may be candidates for treatment of their respiratory failure by ECMO, with access via venovenous (VV) or venoarterial (VA) routes. VV bypass is established with a single cannula through the right internal jugular vein, with blood removed from and infused into the right atrium by separate ports. VA bypass provides additional cardiac support, whereas VV bypass requires a well-functioning heart and relies on the lungs for some oxygenation as well. In VA ECMO, the right atrium is cannulated by means of the internal jugular vein and the aortic arch through the right common carotid artery. As much of the cardiac output is directed through the membrane oxygenator as is necessary to provide oxygenated blood to the infant and remove carbon dioxide. The infant is maintained on bypass until the pulmonary hypertension is resolved and lung function, as measured by compliance and the ability to oxygenate and ventilate, is improved. This is usually seen within 7 to 10 days, but in some infants, it may take up to several weeks to occur. Complications associated with ECMO increase after 14 days and include cannula malposition, bleeding in multiple locations, and infection. The use of ECMO is associated with significant risk. Because patients require systemic anticoagulation, bleeding complications are the most significant. They may occur intracranially or at the site of cannula insertion and can be life threatening. Systemic sepsis is a significant problem and may necessitate decannulation. Criteria for placing infants on ECMO include the presence of normal cardiac anatomy by echocardiography, the absence of fatal chromosome anomalies, and the expectation that the infant would die without ECMO. Traditionally, a threshold of weight greater than 2 kg and gestational age greater than 34 weeks has been applied, although success has been achieved at weights as low as 1.8 kg. Upon decannulation, some centers repair the carotid artery. In instances in which the child is cannulated for a brief period (5 days or less) this may be feasible. A recent study failed to show any benefit from repairing the carotid artery, although this finding remains to be studied further.
A strategy that does not involve the use of ECMO, but instead emphasizes the use of permissive hypercapnia and the avoidance of barotrauma, may provide equal overall outcome in patients with CDH. This likely reflects the fact that mortality is related to the degree of pulmonary hypoplasia and the presence of congenital anomalies, neither of which are correctable by ECMO.
The timing of diaphragmatic hernia repair is still approached by various routes in major centers, particularly when the infant is on ECMO. In patients who are not on ECMO, repair should be performed once the hemodynamic status has been optimized. In neonates who are on ECMO, some surgeons perform early repair on bypass; others wait until the infant’s lungs are improved and the pulmonary hypertension has subsided, repair the diaphragm, and discontinue bypass within hours of surgery. Still others repair the diaphragm only after the infant is off bypass. Operative repair of the diaphragmatic hernia may be accomplished by either an abdominal or transthoracic approach and can be performed either via open or minimally invasive techniques. Through a subcostal incision, the abdominal viscera are withdrawn from the chest, exposing the defect in the diaphragm. Care must be taken when reducing the spleen and liver, as bleeding from these structures can be fatal. The anterior margin is often apparent, while the posterior muscular rim is attenuated. If the infant is heparinized on bypass, minimal dissection of the muscular margins is performed. Electrocautery is used liberally to minimize postoperative bleeding. Most infants who require ECMO support prior to hernia repair have large defects, often lacking the medial and posterior margins. About three fourths of infants repaired on bypass require prosthetic material to patch the defect, suturing it to the diaphragmatic remnant or around ribs or costal cartilages for the large defects. If there is adequate muscle for closure, a single layer of nonabsorbable horizontal mattress sutures, pledgeted or not, closes the defect. Just before the repair is complete, a chest tube may be positioned in the thoracic cavity but is not mandatory. Patients repaired on ECMO are at risk for developing a hemothorax, which can significantly impair ventilation. Anatomic closure of the abdominal wall may be impossible after reduction of the viscera. Occasionally a prosthetic patch or acellular material may be sutured to the fascia to facilitate closure. The patch can be removed at a later time, and the ventral hernia can be closed at that time or subsequently. In patients who are deemed to be candidates for a minimally invasive approach (stable patients, >2 kg, no pulmonary hypertension), a thoracoscopic repair may be safely performed, although concerns have been raised about possible effects of the longer operative time for thoracoscopic repair and higher recurrence rates. If the diaphragm has been repaired on ECMO, weaning and decannulation are accomplished as soon as possible. All infants are ventilated postoperatively to maintain preductal arterial oxygenation of 80 to 100 Torr. Very slow weaning from the ventilator is necessary to avoid recurrent pulmonary hypertension.
Congenital lobar emphysema (CLE) is a condition manifested during the first few months of life as a progressive hyperexpansion of one or more lobes of the lung. It can be life-threatening in the newborn period if extensive lung tissue is involved, but in the older infant and in cases in which the lesion is less severely distended, it causes less respiratory distress. Air entering during inspiration is trapped in the lobe; on expiration, the lobe cannot deflate and progressively overexpands, causing atelectasis of the adjacent lobe or lobes. This hyperexpansion eventually shifts the mediastinum to the opposite side and compromises the other lung. CLE usually occurs in the upper lobes of the lung (left greater than right), followed next in frequency by the right middle lobe, but it also can occur in the lower lobes. It is caused by intrinsic bronchial obstruction from poor bronchial cartilage development or extrinsic compression. Approximately 14% of children with this condition have cardiac defects, with an enlarged left atrium or a major vessel causing compression of the ipsilateral bronchus.
Symptoms range from mild respiratory distress to full-fledged respiratory failure with tachypnea, dyspnea, cough, and late cyanosis. These symptoms may be stationary, or they may progress rapidly or result in recurrent pneumonia. Occasionally, infants with CLE present with failure to thrive, which likely reflects the increased work associated with the overexpanded lung. A hyperexpanded hemithorax on the ipsilateral side is pathognomonic for CLE. Diagnosis is typically confirmed by chest x-ray, which shows a hyperlucent affected lobe with adjacent lobar compression and atelectasis. The mediastinum may be shifted as a consequence of mass effect to the contralateral side causing compression and atelectasis of the contralateral lung (Fig. 39-4). Although chest radiograph is usually sufficient, it is sometimes important to obtain a CT scan of the chest to clearly establish the diagnosis of CLE. This should be done only in the stable patient. Unless foreign body or mucus plugging is suspected as a cause of hyperinflation, bronchoscopy is not advisable because it can lead to more air trapping and cause life-threatening respiratory distress in a stable infant. Treatment is resection of the affected lobe, which can be safely performed using either an open or thoracoscopic approach. Unless symptoms necessitate earlier surgery, resection can usually be performed after the infant is several months of age. The prognosis is excellent.
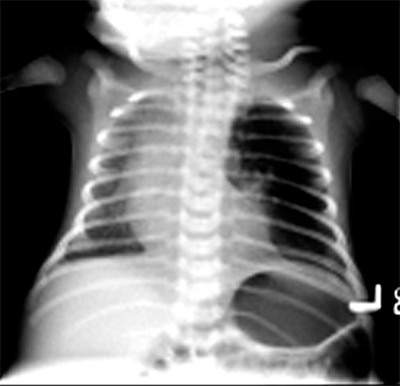
Bronchopulmonary foregut malformations include foregut duplication cysts, congenital pulmonary airway malformations, and pulmonary sequestrations as discussed below.
Previously denoted as congenital cystic adenomatoid malformation (CCAM), congenital pulmonary airway malformation (CPAM) exhibits cystic proliferation of the terminal airway, producing cysts lined by mucus-producing respiratory epithelium, and elastic tissue in the cyst walls without cartilage formation. There may be a single cyst with a wall of connective tissue containing smooth muscle. Cysts may be large and multiple (type I), smaller and more numerous (type II), or resemble fetal lung without macroscopic cysts (type III). CPAMs frequently occur in the left lower lobe. However, this lesion can occur in any location and may occur in more than one lobe on more than one side, although this is rare. Clinical symptoms range from none to severe respiratory failure at birth. Over time, these malformations can be subject to repeated infections and produce fever and cough in older infants and children. The diagnosis is usually confirmed by CT for surgical planning and characteristic features that might delineate other bronchopulmonary foregut malformations (Fig. 39-5). Prenatal US may suggest the diagnosis. Resection is curative and may need to be performed urgently in the infant with severe respiratory distress. Long term, there is a risk of malignant degeneration in unresected CPAMs, but this risk occurs over decades and has not been fully defined. As a result, resection of the affected lobe is usually performed (Fig. 39-6).
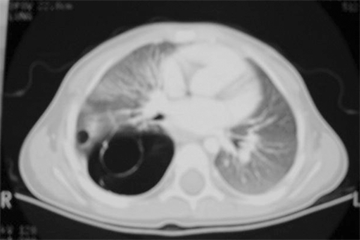
Figure 39-6.
Intraoperative photograph showing left lower lobe congenital pulmonary airway malformation seen in Fig. 39-5.

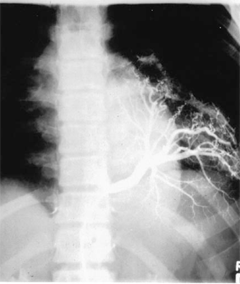
Pulmonary sequestration is uncommon and consists of a mass of lung tissue, usually in the left lower chest, occurring without the usual connections to the pulmonary artery or tracheobronchial tree, yet with a systemic blood supply from the aorta. There are two kinds of sequestration. Extralobar sequestration is usually a small area of nonaerated lung separated from the main lung mass, with a systemic blood supply, located immediately above the left diaphragm. It is commonly found in cases of CDH. Intralobar sequestration more commonly occurs within the parenchyma of the left lower lobe but can occur on the right. There is no major connection to the tracheobronchial tree, but a secondary connection may be established, perhaps through infection or via adjacent intrapulmonary shunts. The blood supply frequently originates from the aorta below the diaphragm; multiple vessels may be present (Fig. 39-7). Venous drainage of both types can be systemic or pulmonary. The cause of sequestration is unknown but most probably involves an abnormal budding of the developing lung that picks up a systemic blood supply and never becomes connected with the bronchus or pulmonary vessels. Sequestrations may, in some cases, exhibit mixed pathology with components consistent with CPAMs. Extralobar sequestration is asymptomatic and is usually discovered incidentally on chest x-ray. If the diagnosis can be confirmed (e.g., by CT scan), resection is not necessary. Diagnosis of intralobar sequestration may be made prenatally and confirmed on postnatal CT scan. Alternatively, the diagnosis of intralobar sequestration may be established after repeated infections manifested by cough, fever, and consolidation in the posterior basal segment of the left lower lobe. Increasingly the diagnosis is being made in the early months of life by US, and color Doppler often can be helpful in delineating the systemic arterial supply. Removal of the entire left lower lobe is usually necessary since the diagnosis often is made late after multiple infections. Occasionally segmental resection of the sequestered part of the lung can be performed using an open or, ideally, a thoracoscopic approach. If an open approach is used, it is important to open the chest through a low intercostal space (sixth or seventh) to gain access to the vascular attachments to the aorta. These attachments may insert into the aorta below the diaphragm; in these cases division of the vessels as they traverse the thoracic cavity is essential. Prognosis is generally excellent. However, failure to obtain adequate control of these vessels may result in their retraction into the abdomen and result in uncontrollable hemorrhage. It is also possible to perform a combined thoracoscopic and open approach, wherein the vessels are clipped and divided thoracoscopically and then the lesion is safely removed through a limited thoracotomy.
Bronchogenic cysts are duplication cysts originating from the airway, regardless of the identity of the epithelial lining. They can occur anywhere along the respiratory tract and can present at any age, although typically they present after accumulation of intraluminal contents, and not within the newborn period. Histologically, they are hamartomatous and usually consist of a single cyst lined with an epithelium; the mesenchyme contains cartilage and smooth muscle. They are probably embryonic rests of foregut origin that have been pinched off from the main portion of the developing tracheobronchial tree and are closely associated in causation with other foregut duplication cysts such as those arising from the esophagus. Bronchogenic cysts may be seen on prenatal US but are discovered most often incidentally on postnatal chest x-ray. Although they may be completely asymptomatic, bronchogenic cysts may produce symptoms, usually compressive in nature, depending on the anatomic location and size, which increase over time if there is no egress of accumulating luminal contents. In the paratracheal region of the neck, they can produce airway compression and respiratory distress. In the lung parenchyma, they may become infected and present with fever and cough. In addition they may cause obstruction of the bronchial lumen with distal atelectasis and infection, or they may cause mediastinal compression. Rarely, rupture of the cyst can occur. Chest x-ray usually shows a dense mass, and CT scan or magnetic resonance imaging (MRI) delineates the precise anatomic location of the lesion. Treatment consists of resection of the cyst, which may need to be undertaken in emergency circumstances for airway or cardiac compression. Resection can be performed either as an open procedure or, more commonly, using a thoracoscopic approach. If resection of a common wall will result in injury to the airway, resection of the inner epithelial cyst lining after marsupialization is acceptable.
Bronchiectasis is an abnormal and irreversible dilatation of the bronchi and bronchioles associated with chronic suppurative disease of the airways. Usually patients have an underlying congenital pulmonary anomaly, cystic fibrosis, or immunologic deficiency. Bronchiectasis can also result from chronic infection secondary to a neglected bronchial foreign body. The symptoms include a chronic cough, often productive of purulent secretions, recurrent pulmonary infection, and hemoptysis. The diagnosis is suggested by a chest x-ray that shows increased bronchovascular markings in the affected lobe. Chest CT delineates bronchiectasis with excellent resolution. The preferred treatment for bronchiectasis is medical, consisting of antibiotics, postural drainage, and bronchodilator therapy, since many children with the disease show signs of airflow obstruction and bronchial hyperresponsiveness. Lobectomy or segmental resection is indicated for localized disease that has not responded appropriately to medical therapy. In severe cases, lung transplantation may be required to replace the terminally damaged, septic lung.
The inherent curiosity of children and their innate propensity to place new objects into their mouths to fully explore them place them at great risk for aspiration. Aspirated objects can be found either in the airway or in the esophagus; in both cases, the results can be life-threatening.
Aspiration of foreign bodies most commonly occurs in the toddler age group. Peanuts are the most common object that is aspirated, although other materials (popcorn, for instance) may also be involved. A solid foreign body often will cause air trapping, with hyperlucency of the affected lobe or lung seen especially on expiration. Oil from the peanut is very irritating and may cause pneumonia. Delay in diagnosis can lead to atelectasis and infection. The most common anatomic location for a foreign body is the right main stem bronchus or the right lower lobe. The child usually will cough or choke while eating but may then become asymptomatic. Total respiratory obstruction with tracheal foreign body may occur; however, respiratory distress is usually mild if present at all. A unilateral wheeze is often heard on auscultation. This wheeze often leads to an inappropriate diagnosis of “asthma” and may delay the correct diagnosis for some time. Chest x-ray will show a radiopaque foreign body, but in the case of nuts, seeds, or plastic toy parts, the only clue may be hyperexpansion of the affected lobe on an expiratory film or fluoroscopy. Bronchoscopy confirms the diagnosis and allows removal of the foreign body. It can be a very simple procedure, or it may be extremely difficult, especially with a smooth foreign body that cannot be grasped easily or one that has been retained for some time. The rigid bronchoscope should be used in all cases, and utilization of the optical forceps facilitates grasping the inhaled object. Epinephrine may be injected into the mucosa when the object has been present for a long period of time, which minimizes bleeding. Bronchiectasis may be seen as an extremely late phenomenon after repeated infections of the poorly aerated lung and may require partial or total resection of the affected lobe. The differential diagnosis of a bronchial foreign body includes an intraluminal tumor (i.e., carcinoid, hemangioma, or neurofibroma).
The most common foreign body in the esophagus is a coin, followed by small toy parts. Toddlers are most commonly affected. The coin is retained in the esophagus at one of three locations: the cricopharyngeus, the area of the aortic arch, or the gastroesophageal junction—all areas of normal anatomic narrowing. Symptoms are variable depending on the anatomic position of the foreign body and the degree of obstruction. There is often a relatively asymptomatic period after ingestion. The initial symptoms are gastrointestinal and include dysphagia, drooling, and dehydration. The longer the foreign body remains in the esophagus with oral secretions unable to transit the esophagus, the greater the incidence of respiratory symptoms including cough, stridor, and wheezing. These findings may be interpreted as signs of upper respiratory infections. Objects that are present for a long period, particularly in children who have underlying neurologic impairment, may manifest as chronic dysphagia. The chest x-ray is diagnostic in the case of a coin. A contrast swallow, or preferably an esophagoscopy, may be required for nonradiopaque foreign bodies. Coins lodged within the upper esophagus for less than 24 hours may be removed using Magill forceps during direct laryngoscopy. For all other situations, the treatment is by esophagoscopy, rigid or flexible, and removal of the foreign body. In the case of sharp foreign bodies such as open safety pins, extreme care is required on extraction to avoid injury to the esophagus. Rarely, esophagotomy is required for removal, particularly of sharp objects. Diligent follow-up is required after removal of foreign bodies, especially batteries, which can cause strictures, and sharp objects, which can injure the underlying esophagus. In the case of a retained battery, this case should be handled as a surgical emergency, as the negative pole of the battery directly damages the surrounding tissue, and tracheoesophageal fistula, aortic exsanguination, and mediastinitis have all been described after local tissue necrosis at the site where the battery has lodged.
ESOPHAGUS
Esophageal atresia (EA) with tracheoesophageal fistula (TEF) is one of the most gratifying pediatric surgical conditions to treat. In the not-so-distant past, nearly all infants born with EA and TEF died. In 1939, Ladd and Leven performed the first successful repair by ligating the fistula, placing a gastrostomy, and reconstructing the esophagus at a later time. Subsequently, Dr. Cameron Haight in Ann Arbor, Michigan, performed the first successful primary anastomosis for EA, which remains the current approach for treatment of this condition. Despite the fact that there are several common varieties of this anomaly and the underlying cause remains obscure, a careful approach consisting of meticulous perioperative care and attention to the technical detail of the operation can result in an excellent prognosis in most cases.
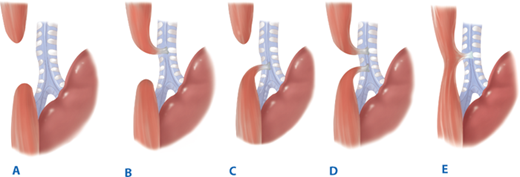
The five major varieties of EA and TEF are shown in Fig. 39-8. The most commonly seen variety is EA with distal TEF (type C), which occurs in approximately 85% of the cases in most series. The next most frequent type is pure EA (type A), occurring in 8% to 10% of patients, followed by TEF without EA (type E). This occurs in 8% of cases and is also referred to as an H-type fistula, based on the anatomic similarity to that letter (Fig. 39-9). EA with fistula between both proximal and distal ends of the esophagus and trachea (type D) is seen in approximately 2% of cases, and type B, EA with TEF between proximal esophagus and trachea, is seen in approximately 1% of all cases.
Figure 39-8.
The five varieties of esophageal atresia and tracheoesophageal fistula. A. Isolated esophageal atresia. B. Esophageal atresia with tracheoesophageal fistula between proximal segment of esophagus and trachea. C. Esophageal atresia with tracheoesophageal fistula between distal esophagus and trachea. D. Esophageal atresia with fistula between both proximal and distal ends of esophagus and trachea. E. Tracheoesophageal fistula without esophageal atresia (H-type fistula).
The esophagus and trachea share a common embryologic origin. At approximately 4 weeks’ gestation, a diverticulum forms off the anterior aspect of the proximal foregut in the region of the primitive pharynx. This diverticulum extends caudally with progressive formation of the laryngotracheal groove, thus creating a separate trachea and esophagus. Successful development of these structures is the consequence of extremely intricate interplay of growth and transcription factors necessary for rostral-caudal and anterior-posterior specification. The variations in clinically observed EA and TEF that must result in failure of successful formation of these structures are depicted in Fig. 39-8. Although definitive genetic mutations have been difficult to identify in isolated EA-TEF, mutations in N-myc, Sox2, and CHD7 have been characterized in syndromic EA-TEF with associated anomalies.
Other congenital anomalies commonly occur in association with EA-TEF. For instance, VACTERRL syndrome is associated with vertebral anomalies (absent vertebrae or hemivertebrae) and anorectal anomalies (imperforate anus), cardiac defects, tracheoesophageal fistula, renal anomalies (renal agenesis, renal anomalies), and radial limb hyperplasia. In nearly 20% of the infants born with EA, some variant of congenital heart disease occurs.
The anatomic variant of infants with EA-TEF predicts the clinical presentation. When the esophagus ends either as a blind pouch or as a fistula into the trachea (as in types A, B, C, or D), infants present with excessive drooling, followed by chocking or coughing immediately after feeding is initiated as a result of aspiration through the fistula tract. As the neonate coughs and cries, air is transmitted through the fistula into the stomach, resulting in abdominal distention. As the abdomen distends, it becomes increasingly more difficult for the infant to breathe. This leads to further atelectasis and respiratory compromise. In patients with type C and D varieties, the regurgitated gastric juice passes through the fistula where it collects in the trachea and lungs and leads to a chemical pneumonitis, which further exacerbates the pulmonary difficulties. In many instances, the diagnosis is actually made by the nursing staff who attempt to feed the baby and notice the accumulation of oral secretions.
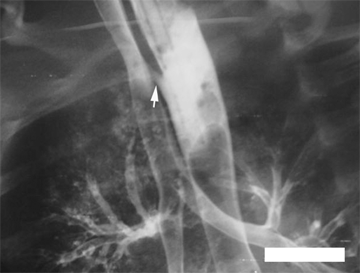
The diagnosis of EA is confirmed by the inability to pass an orogastric tube into the stomach (Fig. 39-10). The dilated upper pouch may be occasionally seen on a plain chest radiograph. If a soft feeding tube is used, the tube will coil in the upper pouch, which provides further diagnostic certainty. An important alternative diagnosis that must be considered when an orogastric tube does not enter the stomach is that of an esophageal perforation. This problem can occur in infants after traumatic insertion of a nasogastric or orogastric tube. In this instance, the perforation classically occurs at the level of the piriform sinus, and a false passage is created, which prevents the tube from entering the stomach. Whenever there is any diagnostic uncertainty, a contrast study will confirm the diagnosis of EA and occasionally document the TEF. The presence of a TEF can be demonstrated clinically by finding air in the gastrointestinal tract. This can be proven at the bedside by percussion of the abdomen and confirmed by obtaining a plain abdominal radiograph. Occasionally, a diagnosis of EA-TEF can be suspected prenatally on US evaluation. Typical features include failure to visualize the stomach and the presence of polyhydramnios. These findings reflect the absence of efficient swallowing by the fetus.
In a child with EA, it is important to identify whether coexisting anomalies are present. These include cardiac defects in 38%, skeletal defects in 19%, neurologic defects in 15%, renal defects in 15%, anorectal defects in 8%, and other abnormalities in 13%. Examination of the heart and great vessels with echocardiography is important to exclude cardiac defects, as these are often the most important predictors of survival in these infants. The echocardiogram also demonstrates whether the aortic arch is left sided or right sided, which may influence the approach to surgical repair. Vertebral anomalies are assessed by plain radiography, and a spinal US is obtained if any are detected. A patent anus should be confirmed clinically. The kidneys in a newborn may be assessed clinically by palpation. An US of the abdomen will demonstrate the presence of renal anomalies, which should be suspected in the child who fails to make urine. The presence of extremity anomalies is suspected when there are missing digits and confirmed by plain radiographs of the hands, feet, forearms, and legs. Rib anomalies may also be present. These may include the presence of a thirteenth rib.
The initial treatment of infants with EA-TEF includes attention to the respiratory status, decompression of the upper pouch, and appropriate timing of surgery. Because the major determinant of poor survival is the presence of other severe anomalies, a search for other defects including congenital cardiac disease is undertaken in a timely fashion. The initial strategy after the diagnosis is confirmed is to place the neonate in an infant warmer with the head elevated at least 30°. A sump catheter is placed in the upper pouch on continuous suction. Both of these strategies are designed to minimize the degree of aspiration from the esophageal pouch. When saliva accumulates in the upper pouch and is aspirated into the lungs, coughing, bronchospasm, and desaturation episodes can occur, which may be minimized by ensuring the patency of the sump catheter. IV antibiotic therapy is initiated, and warmed electrolyte solution is administered. Where possible, the right upper extremity is avoided as a site to start an IV line, as this location may interfere with positioning of the patient during the surgical repair.
The timing of repair is influenced by the stability of the patient. Definitive repair of the EA-TEF is rarely a surgical emergency. If the child is hemodynamically stable and is oxygenating well, definitive repair may be performed within 1 to 2 days after birth. This allows for a careful determination of the presence of coexisting anomalies and for selection of an experienced anesthetic team.
The ventilated, premature neonate with EA-TEF and associated hyaline membrane disease represents a patient who may develop severe, progressive, cardiopulmonary dysfunction. The TEF can worsen the fragile pulmonary status as a result of recurrent aspiration through the fistula and increased abdominal distention, which impairs lung expansion. Moreover, the elevated airway pressure that is required to ventilate these patients can worsen the clinical course by forcing air through the fistula into the stomach, thereby exacerbating the degree of abdominal distention and compromising lung expansion. In this situation, the first priority is to minimize the degree of positive pressure needed to adequately ventilate the child. This can be accomplished using HFOV. If the gastric distention becomes severe, a gastrostomy tube should be placed. This procedure can be performed at the bedside under local anesthetic, if necessary. The dilated, air-filled stomach can easily be accessed through an incision in the left upper quadrant of the abdomen. Once the gastrostomy tube is placed and the abdominal pressure is relieved, the pulmonary status can paradoxically worsen. This is because the ventilated gas may pass preferentially through the fistula, which is the path of least resistance, and bypass the lungs, thereby worsening the hypoxemia. To correct this problem, the gastrostomy tube may be placed under water seal, elevated, or intermittently clamped. If these maneuvers are to no avail, ligation of the fistula may be required. This procedure can be performed in the neonatal intensive care unit if the infant is too unstable to be transported to the operating room. These interventions allow for the infant’s underlying hyaline membrane disease to improve, for the pulmonary secretions to clear, and for the infant to reach a period of stability so that definitive repair can be performed.
In a stable infant, definitive repair is achieved through performance of a primary esophago-esophagostomy. There are two approaches to this operation: open thoracotomy or thoracoscopy. In the open approach, the infant is brought to the operating room, intubated, and placed in the lateral decubitus position with the right side up in preparation for right posterolateral thoracotomy. If a right-sided arch was determined previously by echocardiography, consideration is given to performing the repair through the left chest, although most surgeons believe that the repair can be performed safely from the right side as well. Bronchoscopy may be performed to exclude the presence of additional, upper pouch fistulae in cases of EA (i.e., differentiation of type B, C, and D variants) and to identify a laryngotracheoesophageal cleft.
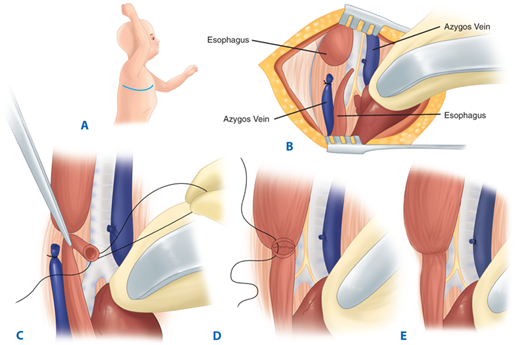
The operative technique for primary repair is as follows (Fig. 39-11). A retropleural approach is generally used, as this technique prevents widespread contamination of the thorax if a postoperative anastomotic leak occurs. The sequence of steps is as follows: (a) mobilization of the pleura to expose the structures in the posterior mediastinum; (b) division of the fistula and closure of the tracheal opening; (c) mobilization of the upper esophagus sufficiently to permit an anastomosis without tension and to determine whether a fistula is present between the upper esophagus and the trachea (forward pressure by the anesthesia staff on the sump drain in the pouch can greatly facilitate dissection at this stage of the operation; care must be taken when dissecting posteriorly to avoid violation of the lumen of the trachea and esophagus); (d) mobilization of the distal esophagus (this needs to be performed judiciously to avoid devascularization, since the blood supply to the distal esophagus is segmental from the aorta; most of the esophageal length is obtained from mobilizing the upper pouch, since the blood supply travels via the submucosa from above); (e) performing a primary esophagoesophageal anastomosis (most surgeons perform this procedure in a single layer using 5-0 sutures; if there is excess tension, the muscle of the upper pouch can be circumferentially incised without compromising blood supply to increase its length; many surgeons place a transanastomotic feeding tube in order to institute feeds in the early postoperative period); and (f) placement of a retropleural drain and closure of the incision in layers.
Figure 39-11.
Primary repair of type C tracheosophageal fistula. A. Right thoracotomy incision. B. Azygos vein transected, proximal and distal esophagus demonstrated, and fistula identified. C. Tracheoesophageal fistula transected and defect in trachea closed. D. End-to-end anastomosis between proximal and distal esophagus (posterior row). E. Completed anastomosis.
When a minimally invasive approach is selected, the patient is prepared for right-sided, transthoracic thoracoscopic repair. The same steps as described earlier for the open repair are undertaken, and the magnification and superb optics that are provided by the thoracoscopic approach provide for superb visualization. Identification of the fistula is performed as a first step; this can be readily ligated and divided between thoracoscopically placed sutures. The anastomosis is performed in a single layer. The thoracoscopically performed TEF repair requires clear and ongoing communication between the operating surgeons and the anesthesiologist; visualization can be significantly reduced with sudden changes in lung inflation, potentially leading to the need to convert to an open repair. Although clear guidelines for patient selection for a thoracoscopic repair as opposed to an open repair remain lacking, reasonable selection criteria include patients over 2.5 kg who are hemodynamically stable and without comorbidities.
The postoperative management strategy of patients with EA-TEF is influenced to a great degree by the preference of the individual surgeon and the institutional culture. Many surgeons prefer not to leave the infants intubated postoperatively, to avoid the effects of positive pressure on the site of tracheal closure. However, early extubation may not be possible in babies with preoperative lung disease either from prematurity or pneumonia or when there is any vocal cord edema. When a transanastomotic tube is placed, feeds are begun slowly in the postoperative period. Some surgeons institute parenteral nutrition for several days, using a central line. The retropleural drain is assessed daily for the presence of saliva, indicating an anastomotic leak. Many surgeons obtain a contrast swallow 1 week after repair to assess the caliber of the anastomosis and to determine whether a leak is present. If there is no leak, feedings are started. The principal benefit of the thoracoscopic approach is that postoperative pain is significantly reduced, as is the requirement for postoperative narcotic analgesia.
Anastomotic leak occurs in 10% to 15% of patients and may be seen either in the immediate postoperative period or after several days. Early leakage (i.e., within the first 24 to 48 hours) is manifested by a new pleural effusion, pneumothorax, and sepsis, and requires immediate exploration. In these circumstances, the anastomosis may be completely disrupted, possibly due to excessive tension. Revision of the anastomosis may be possible. If not, cervical esophagostomy and gastrostomy placement are required, with a subsequent procedure to re-establish esophageal continuity. Anastomotic leakage that is detected after several days usually heals without intervention, particularly if a retropleural approach is used. Under these circumstances, broad-spectrum antibiotics, pulmonary toilet, and optimization of nutrition are important. After approximately a week or so, a repeat esophagram should be performed, at which time the leakage may have resolved.
Strictures are not infrequent (10%–20%), particularly if a leak has occurred. A stricture may become apparent at any time, from the early postoperative period to months or years later. It may present as choking, gagging, or failure to thrive, but often becomes clinically apparent with the transition to eating solid food. A contrast swallow or esophagoscopy is confirmatory, and simple dilatation is usually corrective. Occasionally, repeated dilatations are required. These may be performed in a retrograde fashion, during which a silk suture is placed into the oropharynx and delivered from the esophagus through a gastrostomy tube. Tucker dilators are then tied to the suture and passed in a retrograde fashion from the gastrostomy tube and delivered out of the oropharynx. Increasing sizes are used, and the silk is replaced at the end of the procedure where it is taped to the side of the face at one end and to the gastrostomy tube at the other. Alternatively, image-guided balloon dilation over a guide wire may be performed, using intraoperative contrast radiography to determine the precise location of the stricture and to assess the immediate response to the dilation.
“Recurrent” TEF may represent a missed upper pouch fistula or a true recurrence. This may occur after an anastomotic disruption, during which the recurrent fistula may heal spontaneously. Otherwise, reoperation may be required. Recently, the use of fibrin glue has been successful in treating recurrent fistulas, although long-term follow-up is lacking.
Gastroesophageal reflux commonly occurs after repair of EA-TEF, potentially due to alterations in esophageal motility and the anatomy of the gastroesophageal junction. The clinical manifestations of such reflux are similar to those seen in other infants with primary gastroesophageal reflux disease. A loose antireflux procedure, such as a Nissen fundoplication, is used to prevent further reflux, but the child may have feeding problems after antireflux surgery as a result of the intrinsic dysmotility of the distal esophagus. The fundoplication may be safely performed laparoscopically in experienced hands, although care should be taken to ensure that the wrap is not excessively tight.
Patients with type E TEFs (also called H-type) most commonly present beyond the newborn period. Presenting symptoms include recurrent chest infections, bronchospasm, and failure to thrive. The diagnosis is suspected using barium esophagography and confirmed by endoscopic visualization of the fistula. Surgical correction is generally possible through a cervical approach with concurrent placement of a balloon catheter across the fistula and requires mobilization and division of the fistula. Outcome is usually excellent.
Patients with duodenal atresia and EA-TEF may require urgent treatment due to the presence of a closed obstruction of the stomach and proximal duodenum. In stable patients, treatment consists of repair of the esophageal anomaly and correction of the duodenal atresia if the infant is stable during surgery. If not, a staged approach should be used consisting of ligation of the fistula and placement of a gastrostomy tube. Definitive repair can then be performed at a later point in time.
Primary EA (type A) represents a challenging problem, particularly if the upper and lower ends are too far apart for an anastomosis to be created. Under these circumstances, treatment strategies include placement of a gastrostomy tube and performing serial bougienage to increase the length of the upper pouch. This occasionally allows for primary anastomosis to be performed. Occasionally, when the two ends cannot be brought safely together, esophageal replacement is required using a gastric pull-up, reverse gastric tube, or colon interposition (see below).
Various classification systems have been used to predict survival in patients with EA-TEF and to stratify treatment. A system devised by Waterston in 1962 was used to stratify neonates based on birth weight, the presence of pneumonia, and the identification of other congenital anomalies. In response to advances in neonatal care, the surgeons from the Montreal Children’s Hospital proposed a new classification system. In the Montreal experience, only two characteristics independently affected survival: preoperative ventilator dependence and associated major anomalies. Pulmonary disease as defined by ventilator dependence appeared to be more accurate than pneumonia. When the two systems were compared, the Montreal system more accurately identified children at highest risk. Spitz and colleagues analyzed risk factors in infants who died with EA-TEF. Two criteria were found to be important predictors of outcome: birth weight less than 1500 g and the presence of major congenital cardiac disease. A new classification for predicting outcome in EA was therefore proposed: group I: birth weight ≥1500 g, without major cardiac disease, survival 97% (283 of 293); group II: birth weight <1500 g, or major cardiac disease, survival 59% (41 of 70); and group III: birth weight <1500 g, and major cardiac disease, survival 22% (2 of 9).
In general, surgical correction of EA-TEF leads to a satisfactory outcome with nearly normal esophageal function in most patients. Overall survival rates of greater than 90% have been achieved in patients classified as stable in all the various staging systems. Unstable infants have an increased mortality (40% to 60% survival) because of potentially fatal associated cardiac and chromosomal anomalies or prematurity. However, the use of a staged procedure also has increased survival in even these high-risk infants.
Injury to the esophagus after ingestion of corrosive substances most commonly occurs in the toddler age group. Both strong alkali and strong acids produce injury by liquefaction or coagulation necrosis, and since all corrosive agents are extremely hygroscopic, the caustic substance will cling to the esophageal epithelium. Subsequent strictures occur at the anatomic narrowed areas of the esophagus, cricopharyngeus, midesophagus, and gastroesophageal junction. A child who has swallowed an injurious substance may be symptom free but usually will be drooling and unable to swallow saliva. The injury may be restricted to the oropharynx and esophagus or may extend to include the stomach. There is no effective immediate antidote. Diagnosis is by careful physical examination of the mouth and endoscopy with a flexible or a rigid esophagoscope. It is important to endoscope only to the first level of the burn in order to avoid perforation. Early barium swallow may delineate the extent of the mucosal injury. It is important to realize that the esophagus may be burned without evidence of injury to the mouth. Although previously used routinely, steroids have not been shown to alter stricture development or modify the extent of injury. Therefore, they are no longer part of the management of caustic injuries. Antibiotics are administered during theacute period.
The extent of injury is graded endoscopically as either mild, moderate, or severe (grade I, II, or III). Circumferential esophageal injuries with necrosis have an extremely high likelihood of stricture formation. These patients should undergo placement of a gastrostomy tube once clinically stable. A string should be inserted through the esophagus either immediately or during repeat esophagoscopy several weeks later. When established strictures are present (usually 3–4 weeks), dilatation is performed. Retrograde dilatations are safest, using graduated dilators brought through the gastrostomy and advanced into the esophagus via the transesophageal string. For less severe injuries, dilatation may be attempted in antegrade fashion by either graded bougies or balloons. Management of esophageal perforation during dilation should include antibiotics, irrigation, and closed drainage of the thoracic cavity to prevent systemic sepsis. When recognition is delayed or if the patient is systemically ill, esophageal diversion may be required with staged reconstruction at a later time.
Although the native esophagus can be preserved in most cases, severe stricture formation that does not respond to dilation is best managed by esophageal replacement. The most commonly used options for esophageal substitution are the colon (right colon or transverse/left colon) and the stomach (gastric tube or gastric pull-up). Pedicled or free grafts of the jejunum are less commonly used. The right colon is based on a pedicle of the middle colic artery, and the left colon on a pedicle of the middle colic or left colic artery. Gastric tubes are fashioned from the greater curvature of the stomach based on the pedicle of the left gastroepiploic artery. When the entire stomach is used, as in gastric pull-up, the blood supply is provided by the right gastric artery. The neoesophagus may traverse: (a) substernally; (b) through a transthoracic route; or (c) through the posterior mediastinum to reach the neck. A feeding jejunostomy is placed at the time of surgery, and tube feedings are instituted once the postoperative ileus has resolved. In a recent review of patients treated by gastric pull-up, long-term outcome was very good. Complications included esophagogastric anastomotic leak (n = 15, 36%), which uniformly resolved without intervention, and stricture formation (n = 20, 49%), which responded to a course of dilation. Long-term follow-up has shown that all methods of esophageal substitution can support normal growth and development, and the children enjoy reasonably normal eating habits. Because of the potential for late complications such as ulceration and stricture, follow-up into adulthood is mandatory, but complications appear to diminish with time.
Gastroesophageal reflux (GER) occurs to some degree in all children and refers to the passage of gastric contents into the esophagus. By contrast, gastroesophageal reflux disease (GERD) describes the situation where reflux is symptomatic. Typical symptoms include failure to thrive, bleeding, stricture formation, reactive airway disease, aspiration pneumonia, and apnea. Failure to thrive and pulmonary problems are particularly common in infants with GERD, whereas strictures and esophagitis are more common in older children and adolescents. GERD is particularly problematic in neurologically impaired children.
Because all infants experience occasional episodes of GER to some degree, care must be taken before a child is labeled as having pathologic reflux. A history of repeated episodes of vomiting that interferes with growth and development or the presence of apparent life-threatening events is required for the diagnosis of GERD. In older children, esophageal bleeding, stricture formation, severe heartburn, or the development of Barrett’s esophagus unequivocally connotes pathologic reflux or GERD. In neurologically impaired children, vomiting due to GER must be distinguished from chronic retching.
The workup of patients suspected of having GERD includes documentation of the episodes of reflux and evaluation of the anatomy. A barium swallow should be performed as an initial test. This will determine whether there is obstruction of the stomach or duodenum (due to duodenal webs or pyloric stenosis) and will determine whether malrotation is present. The frequency and severity of reflux should be assessed using a 24-hour pH probe study. Although this test is poorly tolerated, it provides the most accurate determination that GERD is present. Esophageal endoscopy with biopsies may identify the presence of esophagitis and is useful to determine the length of intra-abdominal esophagus and the presence of Barrett’s esophagus. Some surgeons obtain a radioisotope “milk scan” to evaluate gastric emptying, although there is little evidence to show that this test changes management when a diagnosis of GERD has been confirmed using the above modalities.
Most patients with GERD are treated initially by conservative means. In the infant, propping and thickening the formula with rice cereal are generally recommended. Some authors prefer a prone, head-up position. In the infant unresponsive to position and formula changes and the older child with severe GERD, medical therapy is based on gastric acid reduction with an H2-blocking agent and/or a proton pump inhibitor. Medical therapy is successful in most neurologically normal infants and younger children, many of whom will outgrow their need for medications. In certain patients, however, medical treatment does not provide symptomatic relief, and surgery is therefore indicated. The least invasive surgical option includes the placement of a nasojejunal or gastrojejunal feeding tube. Because the stomach is bypassed, food contents do not enter the esophagus, and symptoms are often improved. However, as a long-term remedy, this therapy is associated with several problems. The tubes often become dislodged, acid reflux still occurs, and bolus feeding is generally not possible. Fundoplication provides definitive treatment for GER and is highly effective in most circumstances. The fundus may be wrapped around the distal esophagus either 360° (i.e., Nissen) or to lesser degrees (i.e., Thal or Toupet). At present, the standard approach in most children is to perform these procedures laparoscopically whenever possible. In children with feeding difficulties and in infants under 1 year of age, a gastrostomy tube should be placed at the time of surgery. Early postoperative complications include pneumonia and atelectasis, often due to inadequate pulmonary toilet and pain control with abdominal splinting. Late postoperative complications include wrap breakdown with recurrent reflux, which may require repeat fundoplication, and dysphagia due to a wrap performed too tightly, which generally responds to dilation. These complications are more common in children with neurologic impairment. The keys to successful surgical management of patients with GERD include careful patient selection and meticulous operative technique.
GASTROINTESTINAL TRACT
The majority of infants vomit. Because infant vomiting is so common, it is important to differentiate between normal vomiting, which occurs in almost all babies, to some degree, and abnormal vomiting, which may be indicative of a potentially serious underlying disorder. To determine the seriousness of a particular infant’s bouts of emesis, one needs to characterize what the vomit looks like and how sick the baby is. Vomit that looks like feeds and comes up immediately after a feeding is almost always gastroesophageal reflux. This may or may not be of concern, as described earlier. Vomiting that occurs a short while after feeding or vomiting that projects out of the baby’s mouth may be indicative of pyloric stenosis. By contrast, vomit that has any green color in it is always worrisome. This may be reflective of intestinal volvulus, an underlying infection, or some other cause of intestinal obstruction. A more detailed description of the management of these conditions is provided in the following sections.
Infants with hypertrophic pyloric stenosis (HPS) typically present with nonbilious vomiting that becomes increasingly projectile over the course of several days to weeks due to progressive thickening of the pylorus muscle. HPS occurs in approximately 1 in 300 live births and commonly in infants between 3 and 6 weeks of age. Male-to-female ratio is nearly 5:1.
Eventually, as the pyloric muscle thickening progresses, the infant develops a complete gastric outlet obstruction and is no longer able to tolerate any feeds. Over time, the infant becomes increasingly hungry, unsuccessfully feeds repeatedly, and becomes increasingly dehydrated. Wet diapers become less frequent, and there may even be a perception of less passage of flatus. HPS may be associated with jaundice due to an indirect hyperbilirubinemia, although the nature of this relationship is unclear.
The cause of HPS has not been determined. Studies have shown that HPS is found in several generations of the same family, suggesting a familial link. Administration of erythromycin in early infancy has been linked to the subsequent development of HPS, although the cause is unclear. Infant positioning in the prone position was implicated as a cause of HPS particularly when, in Denmark and Sweden, a drop in both cases of sudden infant death syndrome (SIDS) and HPS were reported. More recently, a similar drop in both SIDS and HPS was noted in Germany, although the distribution of case rate declines were different across regions in Germany, suggesting that the etiology of HPS is not likely simply due to positioning.
Infants with HPS develop a hypochloremic, hypokalemic metabolic alkalosis. The urine pH level is high initially, but eventually drops because hydrogen ions are preferentially exchanged for sodium ions in the distal tubule of the kidney as the hypochloremia becomes severe (paradoxical aciduria). The diagnosis of pyloric stenosis usually can be made on physical examination by palpation of the typical “olive” in the right upper quadrant and the presence of visible gastric waves on the abdomen. When the olive cannot be palpated, US can diagnose the condition accurately in 95% of patients. Criteria for US diagnosis include a channel length of over 16 mm and pyloric thickness over 4 mm.
Given frequent fluid and electrolyte abnormalities at time of presentation, pyloric stenosis is never a surgical emergency. Fluid resuscitation with correction of electrolyte abnormalities and metabolic alkalosis is essential prior to induction of general anesthesia for operation. For most infants, fluid containing 5% dextrose and 0.45% saline with added potassium of 2 to 4 mEq/kg over 24 hours at a rate of approximately 150 to 175 mL/kg per 24 hours will correct the underlying deficit. It is important to ensure that the child has an adequate urine output (>2 cc/kg/h) as further evidence that rehydration has occurred. After resuscitation, a Fredet-Ramstedt pyloromyotomy is performed (Fig. 39-12). It may be performed using an open or laparoscopic approach. The open pyloromyotomy is performed through either an umbilical or a right upper quadrant transverse abdominal incision. The former route is cosmetically more appealing, although the transverse incision provides easier access to the antrum and pylorus. In recent years, the laparoscopic approach has gained great popularity. Two randomized trials have demonstrated that both the open and laparoscopic approaches may be performed safely with equal incidence of postoperative complications, although the cosmetic result is clearly superior with the laparoscopic approach. Whether done through an open or laparoscopic approach, surgical treatment of pyloric stenosis involves splitting the pyloric muscle while leaving the underlying submucosa intact. The incision extends from just proximal to the pyloric vein of Mayo to the gastric antrum; it typically measures between 1 and 2 cm in length. Postoperatively, IV fluids are continued for several hours, after which Pedialyte is offered, followed by formula or breast milk, which is gradually increased to 60 cc every 3 hours. Most infants can be discharged home within 24–48 hours following surgery. Recently, several authors have shown that ad lib feeds are safely tolerated by the neonate and result in a shorter hospital stay.
Figure 39-12.
Fredet-Ramstedt pyloromyotomy. A. Pylorus delivered into wound and seromuscular layer incised. B. Seromuscular layer separated down to submucosal base to permit herniation of mucosa through pyloric incision. C. Cross-section demonstrating hypertrophied pylorus, depth of incision, and spreading of muscle to permit mucosa to herniate through incision.

The complications of pyloromyotomy include perforation of the mucosa (1%–3%), bleeding, wound infection, and recurrent symptoms due to inadequate myotomy. When perforation occurs, the mucosa is repaired with a stitch that is placed to tack the mucosa down and reapproximate the serosa in the region of the tear. A nasogastric tube is left in place for 24 hours. The outcome is generally very good.
The cardinal symptom of intestinal obstruction in the newborn is bilious emesis. Prompt recognition and treatment of neonatal intestinal obstruction can truly be life saving.
The incidence of neonatal intestinal obstruction is 1 in 2000 live births. The approach to intestinal obstruction in the newborn infant is critical for timely and appropriate intervention. When a neonate develops bilious vomiting, one must consider a surgical etiology. Indeed, the majority of newborns with bilious emesis have a surgical condition. In evaluating a potential intestinal obstruction, it is helpful to determine whether the intestinal obstruction is either proximal or distal to the ligament of Treitz. One must conduct a detailed prenatal and immediate postnatal history and a thorough physical examination. In all cases of intestinal obstruction, it is vital to obtain abdominal films in the supine and upright (or lateral decubitus) views to assess the presence of air-fluid levels or free air as well as how far downstream air has managed to travel. Importantly, one should recognize that it is difficult to determine whether a loop of bowel is part of either the small or large intestine, as neonatal bowel lacks clear features, such as haustra or plica circulares, normally present in older children or adults. As such, contrast imaging may be necessary for diagnosis in some instances.
Proximal intestinal obstructions typically present with bilious emesis and minimal abdominal distention. The normal neonate should have a rounded, soft abdomen; in contrast, a neonate with a proximal intestinal obstruction typically exhibits a flat or scaphoid abdomen. On a series of upright and supine abdominal radiographs, one may see a paucity or absence of bowel gas, which normally should be present throughout the gastrointestinal tract within 24 hours. Of utmost importance is the exclusion of a malrotation with midgut volvulus from all other intestinal obstructions as this is a surgical emergency.
Distal obstructions typically presents with bilious emesis and abdominal distention. Passage of black-green meconium should have occurred within the first 24 to 38 hours. Of great importance, one should determine whether or not there is tenderness or discoloration of the abdomen, visible or palpable loops of intestine, and presence or absence of a mass, and whether or not the anus is patent and in the appropriate location. Abdominal radiographs may demonstrate calcifications, which may indicate complicated meconium ileus; pneumatosis and/or pneumoperitoneum may indicate necrotizing enterocolitis. A contrast enema may show whether there is a microcolon indicative of jejunoileal atresia or meconium ileus. If a microcolon is not present, then the diagnoses of Hirschsprung’s disease, small left colon syndrome, or meconium plug syndrome should be considered.
Whenever the diagnosis of duodenal obstruction is entertained, malrotation and midgut volvulus must be excluded. This topic is covered in further detail later in this chapter. Other causes of duodenal obstruction include duodenal atresia, duodenal web, stenosis, annular pancreas, or duodenal duplication cyst. Duodenal obstruction is easily diagnosed on prenatal US, which demonstrates the fluid-filled stomach and proximal duodenum as two discrete cystic structures in the upper abdomen. Associated polyhydramnios is common and presents in the third trimester. In 85% of infants with duodenal obstruction, the entry of the bile duct is proximal to the level of obstruction, such that vomiting is bilious. Abdominal distention is typically not present because of the proximal level of obstruction. In infants with obstruction proximal to the bile duct entry, the vomiting is nonbilious. The classic finding on abdominal radiography is the “double bubble” sign, which represents the dilated stomach and duodenum (Fig. 39-13). In association with the appropriate clinical picture, this finding is sufficient to confirm the diagnosis of duodenal obstruction. However, if there is any uncertainty, particularly when a partial obstruction is suspected, a contrast upper gastrointestinal series is diagnostic.
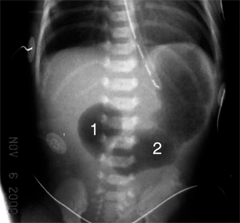
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


