Objectives
- Identify the origin, target organs and cell types, and physiologic effects of parathyroid hormone.
- Describe the functions of osteoblasts and osteoclasts in bone remodeling and the factors that regulate their activities.
- Describe the regulation of parathyroid hormone secretion and the role of the calcium-sensing receptor.
- Identify the sources of vitamin D and describe the biosynthetic pathway involved in modifying it to its biologically active form.
- Identify the target organs and cellular mechanisms of action of vitamin D.
- Describe the negative feedback relationship between parathyroid hormone and the biologically active form of vitamin D.
- Describe the causes and consequences of excess or deficiency of parathyroid hormone and of vitamin D.
- Describe the regulation of calcitonin release and the cell of origin and target organs for calcitonin action.
Parathyroid Gland and Ca2+ and Po4– Regulation: Introduction
The regulation of plasma Ca2+ levels is critical for normal cell function, neural transmission, membrane stability, bone structure, blood coagulation, and intracellular signaling. This regulation relies on the interactions among parathyroid hormone (PTH) from the parathyroid glands, dietary vitamin D, and calcitonin from the thyroid gland (Figure 5–1). PTH stimulates bone resorption and the release of Ca2+ into the circulation. In the kidney, PTH promotes Ca2+ reabsorption and inorganic phosphate excretion in the urine. In addition, PTH stimulates the hydroxylation of 25-hydroxyvitamin D at the 1-position, leading to the formation of the active form of vitamin D (calcitriol). Vitamin D increases intestinal absorption of dietary Ca2+ and facilitates renal reabsorption of filtered Ca2+. In bone, vitamin D increases bone resorption with a resulting increase in the release of Ca2+ into the circulation. Calcitonin counteracts the effects of PTH by inhibiting bone resorption and increasing renal Ca2+ excretion. The overall result of the interactions among PTH, vitamin D, and calcitonin is the maintenance of normal plasma Ca2+ concentrations.
Figure 5–1.
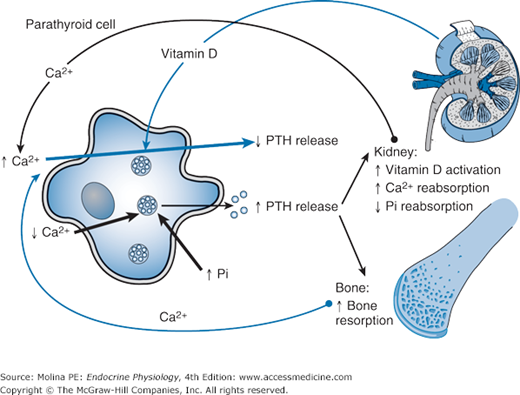
Negative feedback regulation of parathyroid hormone (PTH) release. A sudden decrease in Ca2+ stimulates the release of PTH from the parathyroid gland. PTH increases the activity of 1α-hydroxylase in the kidney, leading to increased activation of vitamin D. In addition, PTH increases the reabsorption of Ca2+ and decreases the reabsorption of inorganic phosphate (Pi). In bone, PTH stimulates bone resorption, increasing the plasma Ca2+ levels. The elevations in vitamin D and plasma Ca2+ levels exert negative feedback inhibition of PTH release. Elevations in plasma Pi levels stimulate the release of PTH.
Functional Anatomy
The parathyroid glands are pea-sized glands located at the top and bottom posterior borders of the lateral lobes of the thyroid gland. The glands are richly vascularized and consist primarily of chief cells, with a thin capsule of connective tissue that divides the gland into lobules. The chief cells synthesize and secrete PTH, a polypeptide hormone that plays a major role in bone remodeling and calcium homeostasis. In addition to its central role in the regulation of Ca2+ levels and bone mass, PTH participates in the renal excretion of phosphate and in the activation of vitamin D. Because of the central role of PTH in calcium homeostasis, its physiologic effects are described in the context of its interaction with the other major regulators of calcium: calcitonin, the hormone synthesized in the C cells of the thyroid gland; and calcitriol, the active form of vitamin D.
Parathyroid Hormone Biosynthesis and Transport
PTH is synthesized as a prepropeptide that is rapidly cleaved to yield pro-PTH and subsequently the mature form of PTH. Mature and intact PTH consists of 84 amino acids. PTH synthesis and release is continuous, with about 6–7 superimposed pulses each hour. PTH is degraded by the kidney and the liver to amino-terminal (PTH 1-34) and carboxy-terminal fragments. The amino-terminal fragments constitute approximately 10% of circulating PTH fragments. They are biologically active but have a short half-life (4–20 minutes). In contrast, the carboxy-terminal fragments, which constitute 80% of the circulating PTH fragments, have no biologic activity and have a longer half-life; they are therefore easier to measure in the plasma. Overall, the intact hormone accounts for 10% of the circulating PTH-related peptides. Because multiple peptide products of PTH breakdown are present in the circulation, determination of the intact molecule is the only reliable index of PTH levels.
 The release of PTH is controlled in a tight feedback system by plasma Ca2+ concentrations. High calcium suppresses PTH secretion and low calcium stimulates hormone release. Small changes in plasma Ca2+ levels are detected by the parathyroid Ca2+-sensing receptor (Figure 5–2). An acute decrease in circulating calcium levels (hypocalcemia) elicits a biphasic wave of PTH release. Preformed PTH is released within seconds, followed by a reduction in intracellular degradation of PTH and increase in PTH release minutes to hours later. Thus, for a given Ca2+ level, there is an optimal PTH level in the circulation. The tight regulation of PTH release by circulating calcium levels is another example of the negative feedback regulation of hormone release. In addition the active form of vitamin D [1,25(OH)2D] also contributes to modulation of PTH levels by reducing PTH gene transcription. Phosphate levels also modulate PTH release, with increased phosphate-stimulating PTH release. Magnesium concentrations also modulate PTH release. Table 5–1 lists the factors that regulate PTH release.
The release of PTH is controlled in a tight feedback system by plasma Ca2+ concentrations. High calcium suppresses PTH secretion and low calcium stimulates hormone release. Small changes in plasma Ca2+ levels are detected by the parathyroid Ca2+-sensing receptor (Figure 5–2). An acute decrease in circulating calcium levels (hypocalcemia) elicits a biphasic wave of PTH release. Preformed PTH is released within seconds, followed by a reduction in intracellular degradation of PTH and increase in PTH release minutes to hours later. Thus, for a given Ca2+ level, there is an optimal PTH level in the circulation. The tight regulation of PTH release by circulating calcium levels is another example of the negative feedback regulation of hormone release. In addition the active form of vitamin D [1,25(OH)2D] also contributes to modulation of PTH levels by reducing PTH gene transcription. Phosphate levels also modulate PTH release, with increased phosphate-stimulating PTH release. Magnesium concentrations also modulate PTH release. Table 5–1 lists the factors that regulate PTH release.
Figure 5–2.
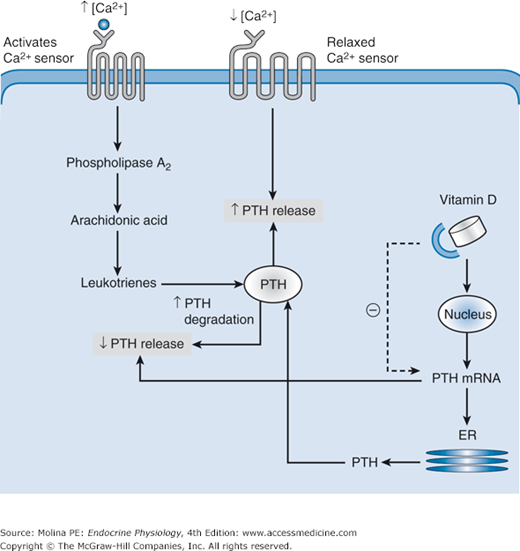
Parathyroid Ca2+-sensing receptor and regulation of parathyroid hormone (PTH) release. Increased Ca2+ levels activate the Ca2+ sensor on parathyroid cells. The calcium sensor is a G protein–coupled receptor that activates phosphatidylinositol–phospholipase C (PI-PLC), leading to intracellular Ca2+ mobilization, protein kinase C (PKC) activation, and downstream activation of phospholipase A2, resulting in activation of the arachidonic acid cascade and the production of biologically active leukotrienes. The leukotrienes trigger the degradation of preformed PTH molecules and decrease the release of intact PTH. The inhibition of PTH release by elevated vitamin D levels is mediated by decreasing the stability of PTH mRNA and consequently the synthesis of PTH. During hypocalcemia, the Ca2+ sensor is in a relaxed conformation and does not activate the second messengers involved in the breakdown of preformed PTH. Acute decreases in plasma calcium cause immediate release of preformed PTH and stimulation of new hormone synthesis. ER, endoplasmic reticulum.
The Ca2+ sensor is a G protein (Gq/11 and Gi)–coupled receptor located on the plasma membrane of the parathyroid chief cells; it is also found in kidney tubule cells and thyroid C cells. The release of PTH is inhibited in response to elevations in plasma Ca2+ concentrations and activation of the Ca2+ receptor (see Figure 5–2). Activation of the Ca2+ receptor initiates a signaling cascade involving phospholipases C, D, and A2. The phosphorylation and activation of phospholipase A2 activate the arachidonic acid cascade and increase leukotriene synthesis. The active leukotriene metabolites inhibit PTH secretion. The inhibition of PTH secretion by elevated Ca2+ levels is the result of increased degradation of preformed hormone stored in secretory granules. The granules are under regulatory control by plasma Ca2+ levels; persistent hypercalcemia leads to rapid degradation of most (90%) of the mature PTH in the cell. This process results in the formation of inactive carboxy-terminal PTH fragments, which are either released into the circulation or further degraded in the parathyroid. The amino acids released during degradation of the formed PTH inside the parathyroid cells are reused in the synthesis of other proteins. In contrast, vitamin D [1,25(OH)2D] inhibits PTH release by decreasing PTH gene expression.
During hypocalcemia, the parathyroid Ca2+ receptor is relaxed, and PTH secretion is not restrained. The rapid secretion of preformed PTH elicited by acute hypocalcemia is rapidly followed by increased stability of PTH mRNA and the synthesis of new hormone.
PTH release is also under regulatory control by phosphate and magnesium levels. Elevations in plasma phosphate levels increase PTH secretion by decreasing phospholipase A2 activity and arachidonic acid formation, thus removing the inhibitory effect on PTH secretion. Elevations in phosphate levels can also affect PTH release indirectly by decreasing plasma Ca2+ levels and vitamin D activation. In contrast, hypophosphatemia markedly decreases PTH mRNA and plasma PTH. The role of phosphate in PTH release is critical in patients with impaired renal function. If not controlled, the increasing levels of phosphate in these individuals result in abnormal elevations in PTH release. As the disease progresses, the negative feedback regulation of PTH release by Ca2+ and vitamin D is also impaired, contributing to the elevation in PTH release. As explained in the following section, chronic elevation of PTH results in excess bone resorption and loss of bone mass.
Plasma Mg2+ concentrations also regulate PTH secretion in a similar manner to that of calcium. PTH release can be stimulated by a moderate decrease in plasma Mg2+. However, very low serum concentrations of Mg2+ induce a paradoxical block in PTH release. The mechanism for this effect has been suggested to be the result of activation of the α-subunits of the calcium-sensing receptor G-proteins, mimicking the activation of the receptor and thus inhibiting PTH secretion.
Plasma magnesium levels reflect those of calcium most of the time. The balance of magnesium is closely linked to that of calcium. Magnesium depletion or deficiency is frequently associated with hypocalcemia. This combined decrease in Mg2+ and Ca2+ leads to impairment in the individual’s ability to secrete PTH. Moreover, severe hypomagnesemia impairs not only the release of PTH from the parathyroid gland in response to hypocalcemia, but it also prevents the responsiveness of bone to PTH-mediated bone resorption. Adrenergic agonists have been shown to increase PTH release through β-adrenergic receptors on parathyroid cells.
Parathyroid Hormone Target Organs and Physiologic Effects
 The primary target organs for the physiologic effects of PTH are kidney and bone. The main physiologic response elicited by PTH is to increase plasma calcium levels by increasing Ca2+ renal reabsorption, Ca2+ mobilization from bone, and intestinal absorption (indirectly via vitamin D3 activation). PTH also increases 1α-hydroxylase activity and renal phosphate excretion. As with other peptide hormones, the effects of PTH are mediated by binding to a cell membrane receptor in target organs. Three types of PTH receptors have been identified (PTHR1, PTHR2, PTHR3), all of which are G protein–coupled receptors. The important physiologic effects of PTH are mediated by PTHR1; the physiologic importance of PTHR2 and PTHR3 is not clear.
The primary target organs for the physiologic effects of PTH are kidney and bone. The main physiologic response elicited by PTH is to increase plasma calcium levels by increasing Ca2+ renal reabsorption, Ca2+ mobilization from bone, and intestinal absorption (indirectly via vitamin D3 activation). PTH also increases 1α-hydroxylase activity and renal phosphate excretion. As with other peptide hormones, the effects of PTH are mediated by binding to a cell membrane receptor in target organs. Three types of PTH receptors have been identified (PTHR1, PTHR2, PTHR3), all of which are G protein–coupled receptors. The important physiologic effects of PTH are mediated by PTHR1; the physiologic importance of PTHR2 and PTHR3 is not clear.
 PTHR1 is expressed in bone osteoblasts and kidney, where it binds PTH and PTH-related protein (PTHrP). PTHrP differs from PTH in structure, sharing only 13 amino acids with the amino-terminal of the hormone, and is a product of a separate gene. PTHrP is important because it mimics the physiologic effects of PTH in the bone and kidney. The identification of PTHrP followed a long search for the factor responsible for the hypercalcemia of malignancy. Because PTHrP is expressed in multiple tissues, it does not mediate its effects in an endocrine fashion, but rather behaves in a paracrine and autocrine manner. PTHrP binds to the PTHR1 in bone and kidney, resulting in elevated plasma Ca2+ levels. Therefore, PTHR1 not only mediates the physiologic effects of PTH, but also plays an important role in the pathophysiologic effects of PTHrP.
PTHR1 is expressed in bone osteoblasts and kidney, where it binds PTH and PTH-related protein (PTHrP). PTHrP differs from PTH in structure, sharing only 13 amino acids with the amino-terminal of the hormone, and is a product of a separate gene. PTHrP is important because it mimics the physiologic effects of PTH in the bone and kidney. The identification of PTHrP followed a long search for the factor responsible for the hypercalcemia of malignancy. Because PTHrP is expressed in multiple tissues, it does not mediate its effects in an endocrine fashion, but rather behaves in a paracrine and autocrine manner. PTHrP binds to the PTHR1 in bone and kidney, resulting in elevated plasma Ca2+ levels. Therefore, PTHR1 not only mediates the physiologic effects of PTH, but also plays an important role in the pathophysiologic effects of PTHrP.
PTH binding to the G protein–coupled PTHR1 initiates a cascade of intracellular processes primarily by signaling through the α-subunit of the stimulatory G-protein (Gαs) leading to an increased synthesis of cyclic 3′,5′-adenosine monophosphate (cAMP) and activation of protein kinase A and phosphorylation of target proteins at serine residues. The result is the activation of preformed proteins, as well as the induction of gene transcription. In addition, activation of the PTHR1 can use additional signaling pathways through Gαq leading to the activation of phospholipase C, increased intracellular inositol 1, 4, 5-trisphosphate and calcium concentrations.
 In the kidney, PTH directly stimulates Ca2+ reabsorption, phosphate excretion, and the activity of 1α-hydroxylase, the enzyme responsible for formation of the active form of vitamin D [1,25(OH)2D]. The site of PTH regulation of Ca2+ reabsorption is the distal tubules. Ca2+ reabsorption by the proximal tubules occurs primarily through a paracellular pathway that is not regulated by hormones or drugs (Figure 5–3). In the thick ascending limbs, Ca2+ is absorbed through a combination of transcellular and paracellular routes. PTH regulates the active, transcellular component, whereas the passive, paracellular route is determined by the extent of concomitant sodium absorption. In the distal tubule, Ca2+ absorption is entirely transcellular and is regulated by PTH, vitamin D, and calcitonin; it can also be affected by Ca2+-sparing drugs such as thiazide diuretics. PTH stimulates the insertion and opening of the apical Ca2+ channel, facilitating the entry of Ca2+ into the cell. These epithelial cells, like the thyroid follicular epithelial cells described in Chapter 4, are polarized; that is, they permit unidirectional flux of the ion from the apical to the basolateral membrane.
In the kidney, PTH directly stimulates Ca2+ reabsorption, phosphate excretion, and the activity of 1α-hydroxylase, the enzyme responsible for formation of the active form of vitamin D [1,25(OH)2D]. The site of PTH regulation of Ca2+ reabsorption is the distal tubules. Ca2+ reabsorption by the proximal tubules occurs primarily through a paracellular pathway that is not regulated by hormones or drugs (Figure 5–3). In the thick ascending limbs, Ca2+ is absorbed through a combination of transcellular and paracellular routes. PTH regulates the active, transcellular component, whereas the passive, paracellular route is determined by the extent of concomitant sodium absorption. In the distal tubule, Ca2+ absorption is entirely transcellular and is regulated by PTH, vitamin D, and calcitonin; it can also be affected by Ca2+-sparing drugs such as thiazide diuretics. PTH stimulates the insertion and opening of the apical Ca2+ channel, facilitating the entry of Ca2+ into the cell. These epithelial cells, like the thyroid follicular epithelial cells described in Chapter 4, are polarized; that is, they permit unidirectional flux of the ion from the apical to the basolateral membrane.
Figure 5–3.
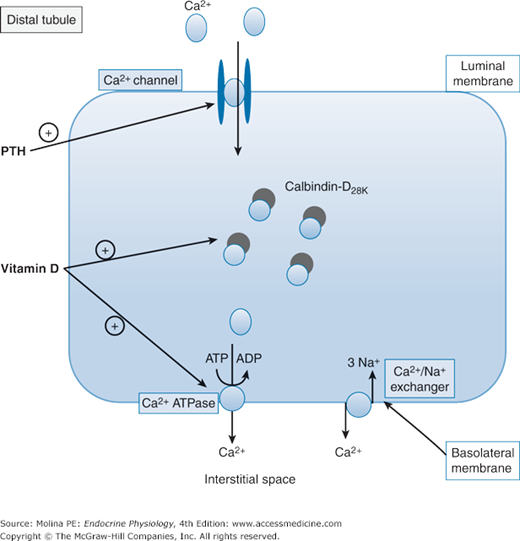
Parathyroid hormone (PTH) effects on renal Ca2+ reabsorption. The transcellular reabsorption of Ca2+ by the distal tubule is regulated by PTH, 1,25(OH)2D, and calcitonin, and by calcium-sparing drugs such as thiazide diuretics. PTH increases the insertion of calcium channels in the apical membrane and facilitates the entry of Ca2+. Inside the cell, Ca2+ binds to calbindin-D28K, a vitamin D-dependent calcium-binding protein that facilitates the cytosolic diffusion of Ca2+ from the apical influx to the basolateral efflux sites. Ca2+ transport out of the cell through the basolateral membrane into the interstitial space is mediated by a Na+/Ca2+ exchanger and a Ca2+– adenosine triphosphatase (ATPase). Vitamin D contributes to the enhanced calcium reabsorption by stimulating the synthesis of calbindin and the activity of Ca2+-ATPase. ADP, adenosine diphosphate; ATP, adenosine triphosphate.
Inside the tubular epithelial cell, Ca2+ binds to calbindin-D28K and then diffuses out through the basolateral membrane (see Figure 5–3). Calbindin-D28K is a vitamin D–dependent calcium-binding protein that is present in the cytosol of cells lining the distal part of the nephron. Calbindin facilitates the cytosolic diffusion of Ca2+ from the apical influx to the basolateral efflux sites. It is thought to act as either a transport protein or a buffer to prevent excess elevation of cytosolic Ca2+ levels. Transport of Ca2+ out of the cell into the interstitial space is mediated by a Na+/Ca2+ exchanger and a Ca2+–adenosine triphosphatase (ATPase). A similar mechanism to aid in Ca2+ absorption is found in intestinal epithelial cells.
 PTH decreases the renal (and intestinal) reabsorption of phosphate by decreasing the expression of the type II Na+/PO42− cotransporter (Figure 5–4). In the kidney, PTH acutely (in minutes to hours) decreases the expression of the Na+/PO42− cotransporter by stimulating their internalization via coated vesicles. PTH binding to its receptor initiates signaling pathways (that are still not well understood) leading to membrane retrieval followed by lysosomal degradation of this transporter. The decreased expression of the transporter, results in decreased phosphate reabsorption. Thus, unlike other transporters such as aquaporin 2 (discussed in Chapter 2), once the Na+/PO42− cotransporter is internalized, it is degraded; thus this is an irreversible internalization. Phosphate reabsorption in the proximal tubule can also be decreased by fibroblast growth factor 23 (FGF23), a peptide produced in osteoblasts and osteocytes. FGF23 suppresses 1α-hydroxylase activity and renal and intestinal Na+/PO42− cotransporter expression (discussed in Chapter 10).
PTH decreases the renal (and intestinal) reabsorption of phosphate by decreasing the expression of the type II Na+/PO42− cotransporter (Figure 5–4). In the kidney, PTH acutely (in minutes to hours) decreases the expression of the Na+/PO42− cotransporter by stimulating their internalization via coated vesicles. PTH binding to its receptor initiates signaling pathways (that are still not well understood) leading to membrane retrieval followed by lysosomal degradation of this transporter. The decreased expression of the transporter, results in decreased phosphate reabsorption. Thus, unlike other transporters such as aquaporin 2 (discussed in Chapter 2), once the Na+/PO42− cotransporter is internalized, it is degraded; thus this is an irreversible internalization. Phosphate reabsorption in the proximal tubule can also be decreased by fibroblast growth factor 23 (FGF23), a peptide produced in osteoblasts and osteocytes. FGF23 suppresses 1α-hydroxylase activity and renal and intestinal Na+/PO42− cotransporter expression (discussed in Chapter 10).
Figure 5–4.
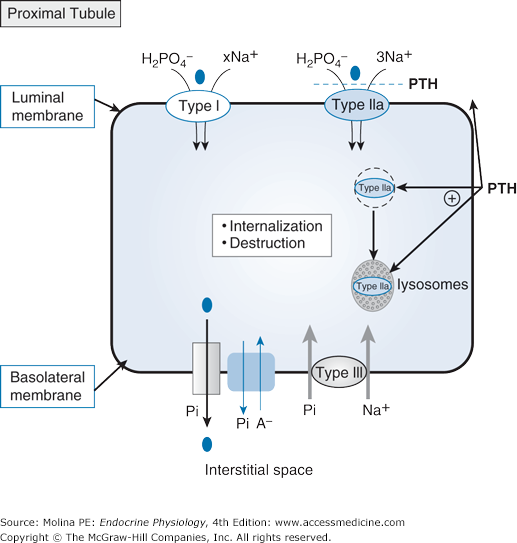
Parathyroid hormone (PTH) effects on renal inorganic phosphate reabsorption. Renal reabsorption of phosphate occurs through apical sodium (Na+)–inorganic phosphate (Pi) cotransport. Three different Na+/Pi cotransporters have been identified: types I, II, and III. Type I and type II cotransporters are located in the apical membrane. Type II cotransporters are expressed in the renal proximal tubule (type IIa) and in the small intestine (type IIb). Type IIa cotransporters are the major target for PTH regulation and contribute to most (up to 70%) of proximal tubular Pi reabsorption. PTH acutely stimulates internalization of the type IIa cotransporters, directing them to the lysosomes for destruction. Type III cotransporters are most likely located at the basolateral membrane and play a general “housekeeping” role in ensuring basolateral Pi influx if apical Pi entry is insufficient to satisfy cellular requirements. Basolateral exit, which is necessary to complete transcellular Pi reabsorption, is not well defined. Several Pi transport pathways have been suggested including Na-Pi cotransport, anion exchange, and even an “unspecific” Pi channel.
 In bone, PTH releases calcium from stores that are readily available and in equilibrium with the extracellular fluid (ECF). Subsequently, PTH stimulates release of calcium (and also phosphate) by activation of bone resorption. Thus, PTH is a major mediator of bone remodeling. PTH binds to receptors found in osteoblasts resulting in a cascade of events culminating in osteoclast activation and leading to a rapid release of Ca2+ from the bone matrix into the extracellular compartment, where it enters the systemic circulation. These receptor-mediated effects of PTH in osteoblasts are mediated through the synthesis or activity of several proteins, including osteoclast-differentiating factor (ODF), also known as receptor activator of nuclear factor-κB ligand (RANKL) or osteoprotegerin ligand (Figure 5–5). PTH-mediated effects in bone involve osteoblast activation and stimulation of genes vital to the processes of degradation of the extracellular matrix and bone remodeling (collagenase-3), production of growth factors (insulin-like growth factor 1), and stimulation and recruitment of osteoclasts (RANKL and interleukin 6). PTH also increases the number of osteoblasts, by decreasing their apoptosis and increasing their proliferation. Although chronic elevations of PTH result in bone resorption, intermittent administration of PTH stimulates bone formation more than bone resorption.
In bone, PTH releases calcium from stores that are readily available and in equilibrium with the extracellular fluid (ECF). Subsequently, PTH stimulates release of calcium (and also phosphate) by activation of bone resorption. Thus, PTH is a major mediator of bone remodeling. PTH binds to receptors found in osteoblasts resulting in a cascade of events culminating in osteoclast activation and leading to a rapid release of Ca2+ from the bone matrix into the extracellular compartment, where it enters the systemic circulation. These receptor-mediated effects of PTH in osteoblasts are mediated through the synthesis or activity of several proteins, including osteoclast-differentiating factor (ODF), also known as receptor activator of nuclear factor-κB ligand (RANKL) or osteoprotegerin ligand (Figure 5–5). PTH-mediated effects in bone involve osteoblast activation and stimulation of genes vital to the processes of degradation of the extracellular matrix and bone remodeling (collagenase-3), production of growth factors (insulin-like growth factor 1), and stimulation and recruitment of osteoclasts (RANKL and interleukin 6). PTH also increases the number of osteoblasts, by decreasing their apoptosis and increasing their proliferation. Although chronic elevations of PTH result in bone resorption, intermittent administration of PTH stimulates bone formation more than bone resorption.
Figure 5–5.
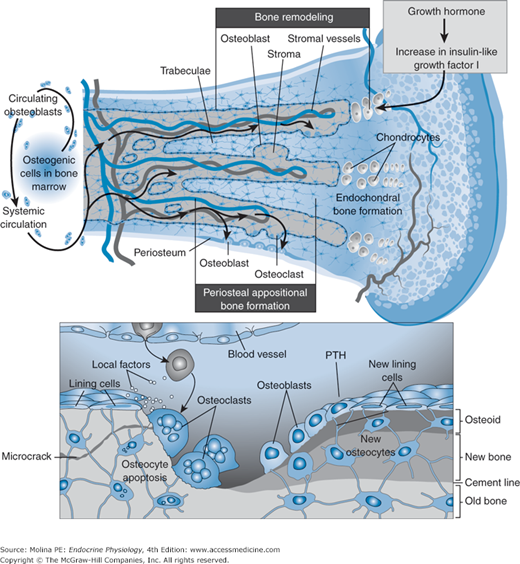
Bone remodeling involves bone formation by osteoblasts and bone resorption by osteoclasts. Parathyroid hormone (PTH) stimulates both aspects of the process. Bone remodeling ensures bone repair and is necessary to maintain calcium homeostasis. (Reproduced with permission from Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357:905–916. Copyright © Massachusetts Medical Society. All rights reserved.)
 To understand how PTH mobilizes calcium from bone, it is important to have a basic idea of bone structure and of the cells involved in mediating the responses to PTH (see Figure 5–5). Bone consists of an extracellular matrix, the organic phase, which is composed of type I collagen, proteoglycans, and noncollagenous proteins. This extracellular bone matrix also contains growth factors and cytokines that have an important regulatory role in bone remodeling, or formation of new bone. The inorganic phase of bone matrix is composed mainly of calcium hydroxyapatite, which functions as a reservoir of calcium and phosphate ions and plays a major role in the homeostasis of these minerals. Most of the skeleton (80%) is composed of cortical bone, found mainly in the shafts of long bones and the surfaces of flat bones. Cortical bone consists of compact bone surrounding central canals (haversian systems) that contain blood vessels, lymphatic tissue, nerves, and connective tissue. Trabecular bone, found mainly at the ends of long bones and within flat bones, consists of interconnecting plates and bars, inside which lies hematopoietic or fatty bone marrow. Three cell types are involved in bone metabolism.
To understand how PTH mobilizes calcium from bone, it is important to have a basic idea of bone structure and of the cells involved in mediating the responses to PTH (see Figure 5–5). Bone consists of an extracellular matrix, the organic phase, which is composed of type I collagen, proteoglycans, and noncollagenous proteins. This extracellular bone matrix also contains growth factors and cytokines that have an important regulatory role in bone remodeling, or formation of new bone. The inorganic phase of bone matrix is composed mainly of calcium hydroxyapatite, which functions as a reservoir of calcium and phosphate ions and plays a major role in the homeostasis of these minerals. Most of the skeleton (80%) is composed of cortical bone, found mainly in the shafts of long bones and the surfaces of flat bones. Cortical bone consists of compact bone surrounding central canals (haversian systems) that contain blood vessels, lymphatic tissue, nerves, and connective tissue. Trabecular bone, found mainly at the ends of long bones and within flat bones, consists of interconnecting plates and bars, inside which lies hematopoietic or fatty bone marrow. Three cell types are involved in bone metabolism.
Osteoblasts express PTH receptors and are responsible for bone formation and mineralization. They are derived from pluripotent mesenchymal stem cells, which can also differentiate into chondrocytes, adipocytes, myoblasts, and fibroblasts. Several hormonal and nonhormonal molecules stimulate the differentiation of osteoblasts from stem cell precursors.
Osteoclasts are large multinucleated bone-resorbing cells derived from hematopoietic precursors of the monocyte-macrophage lineage. They are formed by the fusion of mononuclear cells and are characterized by a ruffled border, which consists of an infolding of the plasma membrane, and a prominent cytoskeleton. Osteoclasts are rich in lysosomal enzymes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


