Papillary Renal Cell Carcinoma
Satish K. Tickoo, MD
Victor E. Reuter, MD
Key Facts
Terminology
Papillary renal cell carcinoma (PRCC)
2nd most common subtype of renal cell carcinoma, usually showing predominant or exclusive papillary architecture, frequently with well-formed tumor capsule
Etiology/Pathogenesis
Majority of sporadic PRCCs are characterized by trisomy of chromosomes 7 and 17, as well as loss of chromosome Y
Clinical Issues
Although majority of patients with unilateral tumors, PRCC is more often bilateral and multifocal compared to other common renal cell tumors
Macroscopic Features
Often surrounded by fibrous pseudocapsule on gross evaluation
Most exhibit variegated cut surface
Microscopic Pathology
Majority of PRCCs exhibit broad morphologic spectrum, including papillary, tubular, and solid patterns
Areas containing papillary architecture seen in most cases
Cores of papillae are mostly loose and fibrovascular, often containing variable amount of foamy macrophages
WHO divides papillary RCC into type 1 and type 2
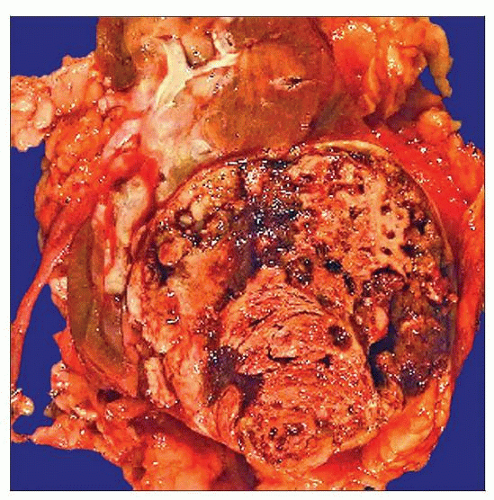 Gross photograph of a papillary RCC shows an encapsulated mass with a necrotic and hemorrhagic cut surface. Among the common renal cell tumors, papillary RCC is the most likely to have a capsule. |
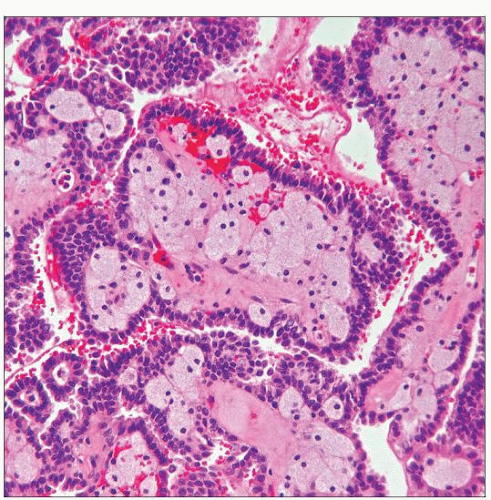 This photomicrograph shows the typical features of a papillary RCC, with prominent papillae and abundant foamy macrophages in the papillary cores. The lining cells are amphophilic and cuboidal. |
TERMINOLOGY
Abbreviations
Papillary renal cell carcinoma (PRCC)
Synonyms
Chromophilic renal cell carcinoma
Definitions
2nd most common subtype of renal cell carcinoma, usually showing predominant or exclusive papillary architecture, frequently with well-formed tumor capsule
ETIOLOGY/PATHOGENESIS
Molecular Characteristics
Majority of sporadic PRCCs are characterized by trisomy of chromosomes 7 and 17, as well as loss of chromosome Y
Trisomies of chromosomes 12, 16, and 20 and loss of heterozygosity at 9p13 are observed in some cases
Some investigators suggest that tumors with trisomy 7/17 alone are likely to have benign behavior
Whereas those with additional genetic abnormalities behave aggressively
More gains of 7p and 17p by CGH reported by some in type 1, compared to type 2 tumors
Activating mutations of MET oncogene, located at 7q31, present in all cases of hereditary papillary renal carcinoma syndrome
Similar mutations observed in approximately 13% sporadic papillary RCCs
CLINICAL ISSUES
Epidemiology
Incidence
Comprise 11-15% of renal cell neoplasms
Age
Ranges from 3rd to 8th decades of life with peak incidence in 6th and 7th decades
Similar to other renal cell tumors
Gender
Reported M:F = 1.8:1-4:1
Presentation
> 50% of cases present as incidental masses, detected on radiologic investigation for unrelated conditions
Reported size ranges from 1-18 cm (median: 6.4 cm)
However, downward size migration seen in modern times due to incidental discovery on imaging
Although majority of patients have unilateral tumors, PRCC is more often bilateral and multifocal compared to other common renal cell tumors
Treatment
Surgical approaches
Partial nephrectomy is preferred option
Total or radical nephrectomy rarely performed now at some institutions even for tumors > 4 cm in size
Drugs
Resistance to systemic therapy characterizes patients with metastatic papillary RCC
Targeted therapies against VEGF tyrosine kinases (e.g., sunitinib) in metastatic PRCC with clinical responses in occasional case
Similar targeted therapies are focus of attention in multiple ongoing clinical trials
Targeting MET signaling pathway is another therapeutic approach under active investigation
Prognosis
Overall, 5- and 10-year survivals better than clear cell RCC, and possibly worse than chromophobe RCC
However, some studies show no significant prognostic differences between PRCC and chromophobe RCC
MACROSCOPIC FEATURES
General Features
Well-circumscribed mass
Often surrounded by fibrous pseudocapsule on gross evaluation
Of all common renal cell tumor types, papillary renal cell carcinoma is most likely to be surrounded by fibrous pseudocapsule
Most exhibit variegated cut surface
Color is related to microscopic findings
Tumors with abundant foamy macrophages, tan to yellow
Tumors with intratumoral hemorrhage, dark tan to brown
Grossly visible areas of necrosis, hemorrhage, and cystic change very common, present in 32-70% of tumors
Some tumors almost entirely necrotic
Multifocality is present in > 45% of cases
In some, this is reported to be only a microscopic finding
Many such microscopic tumors may be considered papillary adenomas now
MICROSCOPIC PATHOLOGY
Histologic Features
Majority of PRCCs exhibit broad morphologic spectrum, including papillary, tubular, and solid patterns
Papillary patterns include
Classic papillary with discrete papillary fronds lined by neoplastic cells with central fibrovascular core
Papillary-trabecular with delicate, elongated papillations arranged in parallel fashion
Papillary-solid with closely packed papillae, sometimes masking their true growth pattern
Areas containing papillary architecture seen in most cases
However, > 50% show variable proportion of “solid,” tubular, &/or glomeruloid growth patterns
Glomeruloid growth pattern composed of tubular structures with intraluminal tufting of tumor cells
Cells lining tubules are cuboidal with scant to moderate amphophilic cytoplasm
Cells tufting into lumen with abundant eosinophilic cytoplasm and usually higher-grade nuclei
Rarely, sarcomatoid growth may be seen and is sign of aggressive disease
Cores of papillae are mostly loose and fibrovascular, often containing variable number of foamy macrophages
However, variations in morphology of cores are not uncommon, and may include
Cores with no macrophages
Cores with branching papillae
Some papillae with no distinct cores, as in tumors with micropapillary features
Marked hyalinization of cores
Variable degree of edema, sometimes leading to fluid-filled, grape-like polypoid structures
WHO divides papillary RCC into 2 types
Type 1 with papillae covered by smaller cells with scant, amphophilic cytoplasm
Type 2 with larger tumor cells, often with higher nuclear grade, eosinophilic cytoplasm, and nuclear pseudostratification
Type 1 tumors more often positive for CK7 than type 2 PRCC
Reportedly worse prognosis in type 2 compared to type 1 tumors
Trisomies 7 and 17 more often reported in type 1 than type 2 PRCC
MET gene mutations only present in type 1 PRCC
Many tumors that are not PRCC and have prominent papillary architecture are more often mistakenly regarded as type 2 PRCC
Psammoma bodies, hemosiderin-laden macrophages, hemosiderin deposition within tumor cells are often seen in PRCC
WHO classification system does not address issue of PRCC with mixture of type 1 and type 2 morphologic features
Combination of features of both types not uncommon
In older literature, such tumors designated as “duophilic”
Recently, tumors with oncocytic cytoplasm with low-grade nonoverlapping nuclei described as “oncocytic” PRCC
Immunohistochemically CK7 positive
Show trisomies 7 and 17
Show biologic behavior similar to type 1 PRCC
Features suggest that eosinophilic PRCC with low-grade nuclei are molecularly and biologically similar to type 1 tumors
Molecular evaluation also suggests modified classification of PRCC
Type 1: Similar to type 1 of WHO
Type 2A: Tumors with eosinophilic cytoplasm but low-grade nuclei
Type 2B: Tumors with mixture of type 1 and type 2A features
Type 2C: Tumors with high-grade nuclei; tumors often with topoisomerase II-α overexpression
Nuclear grading
Many believe Fuhrman grading system is well suited for PRCC
Others disagree and do not use Fuhrman grading scheme in PRCC
Others recently proposed only assessment of nucleolar prominence in most pleomorphic foci rather than Fuhrman grade
Predominant Pattern/Injury Type
Neoplastic
Predominant Cell/Compartment Type
Epithelial
ANCILLARY TESTS
Immunohistochemistry
Diffuse positivity for CK7 very frequent
More often in type 1 than type 2 tumors
AMACR diffusely positive, with cytoplasmic granular staining
CD10 often positive, usually with luminal membranous staining
CA9 either negative or focally positive
Positivity usually limited to papillary tips or perinecrotic areas
RCC and pax-2 positive
DIFFERENTIAL DIAGNOSIS
Clear Cell RCC Exhibiting Papillary or Pseudopapillary Growth
Focal papillary architecture may be seen in clear cell RCC
Usually result of cell drop-off in areas away from feeding vessels
This usually creates pseudopapillary appearance
Adequate sampling and presence of typical cytoarchitectural features of clear cell RCC in other areas should clarify issue
Prominent psammoma bodies and hemosiderin deposition within tumor cells are not present in clear cell RCC
Any fibrovascular cores would be unusual in clear cell RCC and, if present, would be focal
Clear cells in clear cell RCC usually have optically transparent, completely clear cytoplasm
In papillary RCC, clear-appearing cells usually with variable granularity and often fine hemosiderin
CK7 and AMACR immunoreactivity usually (-) or very focal in clear cell RCC
CA9 shows diffuse membranous reactivity in majority of clear cell RCCs
Collecting Duct Carcinoma (CDC)
Has variable amount of papillary growth pattern
Is centered in medulla
Invariably widely infiltrative and pseudocapsule, if present, is focal
Virtually always high grade
Associated with pronounced desmoplastic stroma
Shows prominent multinodular growth pattern and glandular and sheet-like architecture
Intracytoplasmic and luminal mucin, often focal, common feature
Often reactive for CEA, PNA, soybean agglutin, ULEX-1, Ulex europaeus, and HMCK(34βE12)
CK7 may be expressed in both tumors
Cytogenetic studies may be needed to solve difficult diagnostic problems
MiTF/TFE Family Translocation-associated Renal Carcinomas (TFE Carcinoma)
More common in young patients
Rarely multifocal
Admixture of solid, nested, and papillary growth
Usually with high nuclear grade and often with cells showing voluminous cytoplasm
Cytoplasm varies from clear to eosinophilic
(-) or only focally (+) for cytokeratins and EMA/MUC1
Characteristically exhibit nuclear immunoreactivity for TFE3 or TFEB, depending on the translocation
Hereditary Leiomyomatosis RCC (HLRCC)-Related Renal Carcinoma
Tumors show variable, but often prominent, papillary architecture
Considered to be type 2 PRCCs in the past
Other architectural patterns, including glandular, alveolar, and solid are often present
Usually CK7 negative
History of leiomyomas, both uterine and others, is common
Most characteristic morphologic feature is very prominent nucleoli with perinucleolar halos
Molecular evidence of fumarate hydratase (FH) gene alterations
Clear Cell Papillary RCC (CC-PRCC)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


