 FIGURE 11.1. Incidence of papillary carcinoma in the United States in 1973-2008. Based on the SEER data.5 |
of external beam radiation therapy for benign conditions of the head and neck, common in the United States and several other countries in the 1930s to 1940s, resulted in a significant increase of thyroid cancer incidence in individuals exposed during childhood.18,19 However, the use of radiation for treatment of benign conditions was abandoned in the 1950s. As a result, the impact of radiation as an etiologic agent has been gradually decreasing. For example, a history of radiation exposure was documented in 12% of patients with thyroid cancer diagnosed in 1978 to 1980 as compared with 5% of patients diagnosed in 1996.20,21 However, radiation therapy for malignant tumors, such as Hodgkin disease, remains a source of radiation-induced papillary carcinoma. The Chernobyl nuclear power accident in 1986 has lead to the development of papillary carcinomas in >5,000 individuals exposed to radioiodines during childhood and adolescence.22,23,24 The increase in cancer risk due to diagnostic X-ray procedures, occupational radiation exposure, and high dose 131I therapy for hyperthyroidism has not been found.21,25
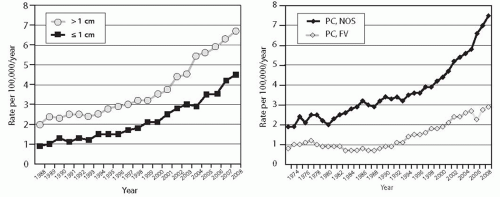 |
 FIGURE 11.3. Mean age of patients with papillary carcinomas in the United States in 1974-2008. Based on the SEER data.5 |
Table 11.1 Radiation Exposure and Thyroid Papillary Carcinoma | |||||||
|---|---|---|---|---|---|---|---|
|
resulting in a valine-to-glutamate replacement at residue 600 (V600E).110,114 BRAF V600E comprises 98% to 99% of all BRAF mutations found in thyroid cancer. Other alterations involve K601E point mutation and small in-frame insertions or deletions surrounding codon 600,115,116,117,118,119 as well as AKAP9/BRAF rearrangement.37 The rearrangement is a paracentric inversion of chromosome 7q leading to the fusion between the portion of BRAF gene encoding the protein kinase domain to the AKAP9 gene.37 All point mutations and the rearrangement lead to the activation of BRAF kinase and chronic stimulation of the MAPK pathway.
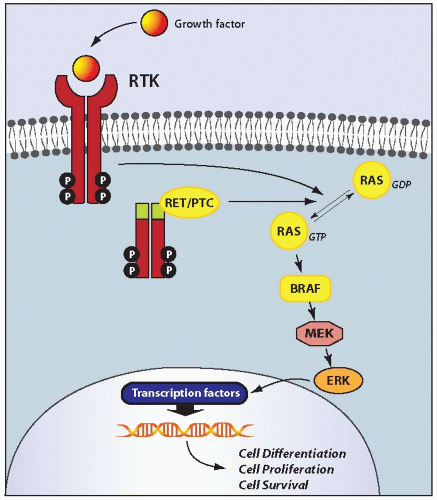 FIGURE 11.4. Schematic representation of the MAPK signaling pathway. Physiologically, binding of growth factors to a receptor tyrosine kinase (RTK), such RET and NTRK1, results in receptor dimerization and activation via autophosphorylation of tyrosine residues in the intracellular domain. The activated receptor through a series of adaptor proteins leads to activation of RAS, located at the inner surface of the plasma membrane, by substitution of GDP with GTP. The active, GTP-bound form of RAS binds to and recruits RAF proteins, mainly BRAF in thyroid follicular cells, to the plasma membrane. Activated BRAF phosphorylates and activates MEK and ERK. Once activated, ERK phosphorylates cytoplasmic proteins and translocates into the nucleus, where it regulates transcription of the genes involved in cell differentiation, proliferation, and survival. Activation of this pathway in papillary carcinoma occurs as a result of point mutation or chromosomal rearrangement affecting the RET, RAS, and BRAF genes. |
Table 11.2 MAPK-activating Mutations in Papillary Carcinomas and Their Clinical and Phenotypical Associations | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 11.3 Types of RET/PTC Rearrangement in Papillary Carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
carcinomas and also in various other thyroid tumors and benign lesions.146
 FIGURE 11.5. Nuclear architecture predisposes to the generation of RET/PTC rearrangements in thyroid cells. Nucleus of normal human thyroid follicular cell hybridized with probes for the RET (green color), H4 (red), and NCOA4 (orange) genes showing close proximity of RET and NCOA4 (fusion partners in RET/PTC3) on one copy of chromosome 10 (upper left) and proximity of RET and H4 (fusion partners in RET/PTC1) on another copy of chromosome 10 (bottom). |
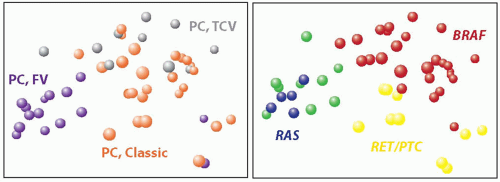 FIGURE 11.6. Correlation between gene expression profiles and histologic variants and mutations in papillary carcinoma. (Left) Principal component analysis based on the expression data for over 22,000 genes shows distinct clusters formed by cases of classic papillary carcinoma (orange spheres), follicular variant (purple), and tall cell variant (grey). (Right) Similar analysis with reference to mutational status demonstrates clusters of tumors carrying BRAF mutation (red), RET/PTC rearrangement (yellow), and RAS mutation (blue). Green spheres represent papillary carcinomas with no mutation. (Based on the data reported by Giordano TJ, Kuick R, Thomas DG, et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene. 2005;24:6646-6656.) |
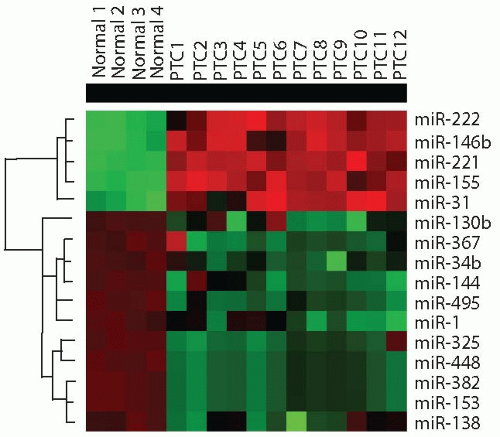 FIGURE 11.7. miRNAs dysregulation in papillary thyroid carcinoma. Cluster dendrogram demonstrates several upregulated (red) and downregulated (green) miR-NAs in papillary carcinomas (PTC) as compared with normal thyroid cells. (Based on data reported by Nikiforova MN, Tseng GC, Steward D, et al. MicroRNA expression profiling of thyroid tumors: biological significance and diagnostic utility. J Clin Endocrinol Metab. 2008;93:1600-1608 and Yip L, Kelly L, Shuai Y, et al. MicroRNA signature distinguishes the degree of aggressiveness of papillary thyroid carcinoma. Ann Surg Oncol. 2011;18:2035-2041.) |
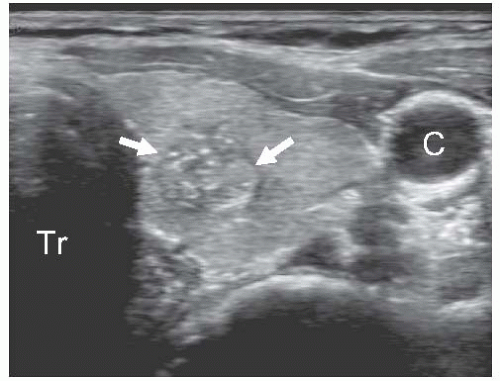 FIGURE 11.8. Ultrasonographic features of papillary carcinoma. Transverse grey scale image of the left thyroid lobe shows an irregularly-shaped isoechoic nodule (arrows) with ill-defined borders and several punctuate hyperechoic foci of microcalcifications, which correspond to psammoma bodies found on microscopic examination. Tr, trachea; C, carotid artery. |
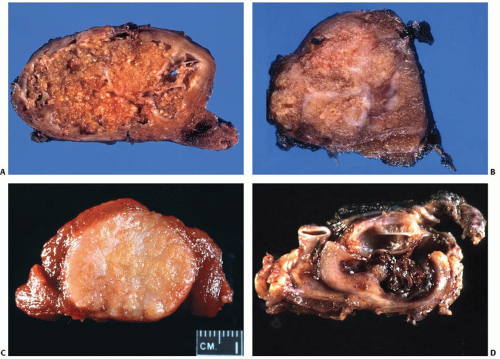 FIGURE 11.9. Gross appearance of papillary carcinoma. A, B: Two examples of classic papillary carcinoma. C: Follicular variant of papillary carcinoma. The tumor is well demarcated and has smooth borders but shows no capsule. D: Metastastatic papillary carcinoma to a lymph node with marked cystic change. |
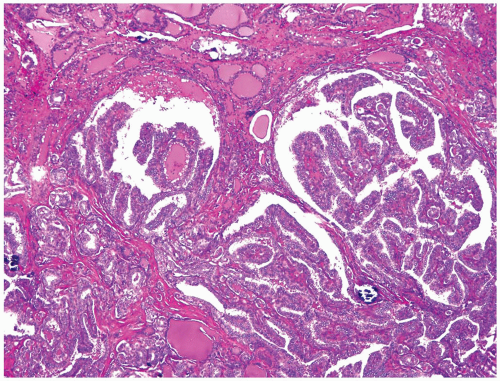 FIGURE 11.10. Low-power view of papillary carcinoma. Note an infiltrative border and a predominantly papillary architecture with occasional neoplastic follicles and scattered calcifications. |
The nuclei exhibit characteristic microscopic changes that serve as a core requirement for the diagnosis of papillary carcinoma. The following main diagnostic nuclear features are recognized:
Table 11.4 Main Microscopic Features of Papillary Carcinoma | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
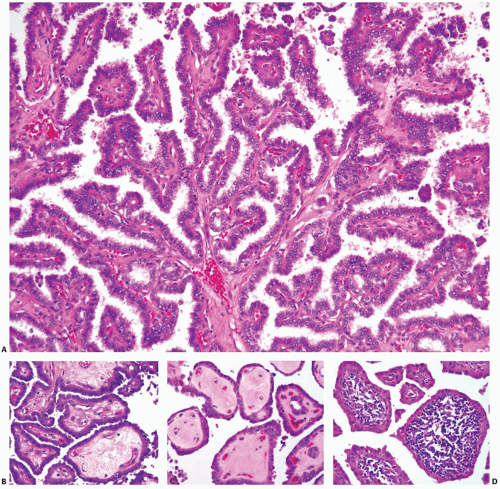 FIGURE 11.11. Papillae of papillary carcinoma. A: Typical tumor papillae with well-developed fibrovascular cores and complex branching. Some papillae may have stalks composed of loose myxoid stroma (B) or acellular hyalinized stroma (C) or contain dense lymphoid infiltration in the stalks (D). |
appearance requires a tissue fixation step, as it is not seen in frozen sections or FNA smears.217 Formalin fixation most consistently yields nuclear clearing, which is also seen after fixation in Bouin fluid, Zenker fluid, or B5.216 However, other fixatives such as SafeFix and HistoChoice tend not to show this nuclear appearance (Lester D. R. Thompson, unpublished data). Although changes in the distribution of chromatin and ribonucleoproteins have been seen in these tumors by electron microscopy,219,220 the exact mechanism of nuclear clearing is not well understood.
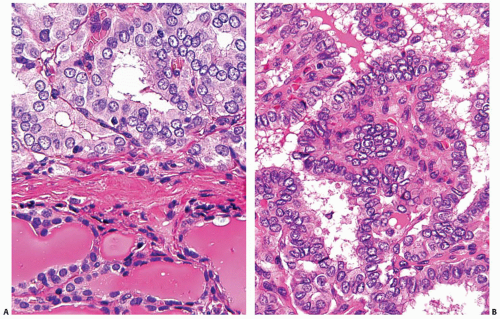 FIGURE 11.12. Nuclear features of papillary carcinoma. A: Enlarged size of tumor nuclei (top) is easier to appreciate when compared with adjacent normal thyroid tissue (bottom). B: Nuclear overlapping and crowding with “lakes” of overlapping nuclei. Many nuclei have finely dispersed chromatin and empty appearance with prominent nuclear membrane due to chromatin margination. |
 FIGURE 11.13. Marked irregularity of the nuclear contours. This important feature of papillary carcinoma can be seen on light microscopy (A) and electron microscopy (B). |
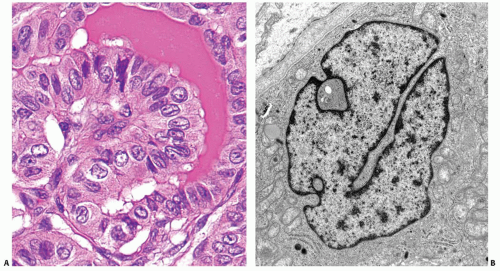 FIGURE 11.14. Nuclear grooves. These structures are often seen in papillary carcinoma cells on light microscopy (A). They are formed by linear invagination of the nuclear membrane that travels deep into the nuclear volume, which is best appreciated on electron microscopy (B). |
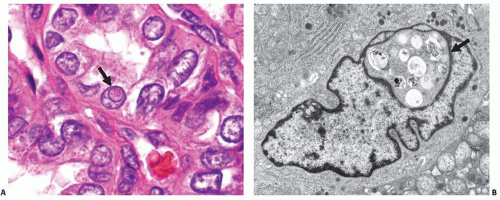 FIGURE 11.15. Nuclear pseudoinclusions. A: Light microscopy image showing a cell in the center of the field that contains an eosinophilic intranuclear body (arrow), which is demarcated by a rim of nuclear membrane and stained more like the cytoplasm than the remainder of the nucleus. B: Ultramicroscopic image of a cell containing a nuclear pseudoinclusion (arrow), which is surrounded by a nuclear membrane and contains phagolysosomes and other cytoplasmic organelles. |
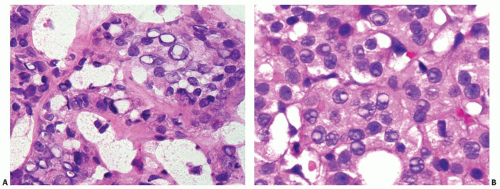 FIGURE 11.16. Structures mimicking nuclear pseudoinclusions. A: An intraoperative frozen section showing numerous nuclei with sharpededged, punched-out empty spaces. B: A routine formalin fixed section showing multiple nuclei with bubbles that are pale stained and lack the texture of the adjacent cytoplasm. Many of the bubbles contain nucleoli. |
have a round or spherical shape,
show concentric layers of calcium deposition, and
be located in association with tumor cells, in the tumor stroma, or in a lymphatic channel, but not within the lumen of a follicle (Fig. 11.17).
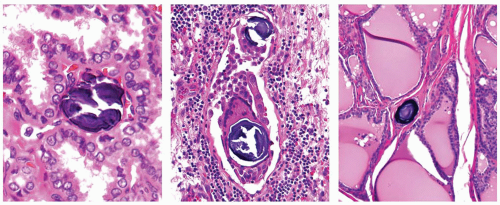 FIGURE 11.17. Psammoma bodies. True psammoma bodies are located either in a stalk of the papillae (left) or in association with tumor cells and multinucleated giant cells within a lymphatic channel (middle), or in isolation within thyroid stroma (right). |
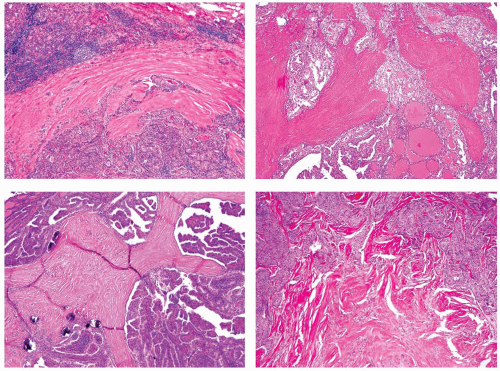 FIGURE 11.18. Various appearances of fibrosis in papillary carcinoma. Significant lymphocytic infiltration is found within the tumor stroma or in thyroid tissue surrounding the tumor in 25% to 40% of cases.113,213,237 Prominent stromal infiltration is a characteristic feature of the Warthin-like variant as described later in the chapter. Infiltration in the adjacent thyroid tissue may be either reactive (peritumor thyroiditis, see Chapter 4) or a part of chronic lymphocytic (Hashimoto) thyroiditis. As peritumoral lymphocytic infiltration is common in papillary carcinoma, this finding should not lead to the diagnosis of chronic lymphocytic (Hashimoto) thyroiditis, unless the same changes are found in thyroid tissue remote from the tumor, preferably in the opposite lobe. |
subcutaneous soft tissues, larynx, trachea, esophagus, or recurrent laryngeal nerve (corresponds to T4) (Fig. 11.20 B). Extrathyroidal extension is found in 20% to 25% of tumors213,240 and is more common in BRAF-positive papillary carcinomas and less common in the follicular variant.113 As discussed later in the chapter, extrathyroidal extension is an important prognostic factor, although some more recent studies suggest that only extensive extrathyroidal invasion correlates with prognosis.247,248
 FIGURE 11.19. Additional changes seen in papillary carcinoma. A: Darker eosinophilic colloid in papillary carcinoma (right) as compared with adjacent thyroid (left). B: Multinucleated giant cell in a space between papillae. C: Two foci of squamous metaplasia. D: Cystic change in papillary carcinoma. |
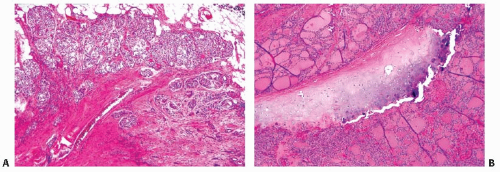 FIGURE 11.20. Extrathyroidal extension. A: Minimal extrathyroidal extension into adjacent soft tissue (TNM, T3). B: Extensive extrathyroidal extension involving tracheal cartilage (TNM, T4a). |
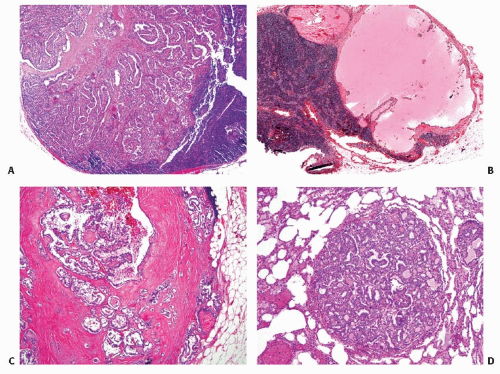 FIGURE 11.21. Papillary carcinoma metastases. A: Tumor metastasis to a lymph node. B: Lymph node metastasis with marked cystic change. C: Lymph node metastasis with tumor extension through the lymph node capsule. D: Tumor metastasis to the lung. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


