KEY POINTS
Incomplete fusion of the dorsal and ventral pancreatic ducts results in pancreas divisum, but a variety of ductal anomalies can be seen. Magnetic resonance cholangiopancreatography as well as endoscopic retrograde cholangiopancreatography can identify these ductal anomalies, and clarification of the ductal pattern of the pancreas is important before attempts at interventions.
The “replaced right hepatic artery” occurs in 15% of patients and needs to be identified preoperatively to prevent inadvertent injury with resulting hepatic necrosis. Anomalous hepatic arterial anatomy can result in hepatic ischemia during dissection of the porta hepatis as well. “Thin cut” multidetector com.puted tomographic images are usually able to identify the relevant arterial and venous patterns around the pancreas.
Regardless of the etiology, the management of the early phase of acute pancreatitis is critical to achieve a successful outcome. Aggressive fluid resuscitation and early enteral feeding both reduce the risk of complications. It is no longer considered appropriate to “rest the pancreas” if the patient can tolerate enteral nutrients.
Surgical intervention in acute pancreatitis is reserved for patients with infected collections or infected necrosis only, or to relieve an impacted gallstone in the ampulla if endoscopic or radiologic treatments are unavailable or unsuccessful. Infection is usually confirmed by a pattern of air in the retroperitoneum on computed tomographic scan, or by documentation of bacteria on Gram’s stain or culture from fine-needle aspiration of a suspected infected fluid collection. Fine-needle aspiration of suspicious fluid collections should not be converted to percutaneous drainage unless infection is confirmed, and the consensus decision has been made that percutaneous drainage is appropriate for the individual patient.
The appearance of chronic pancreatitis on computed tomographic scan varies dramatically, and multiple diagnostic studies are usually needed to establish the extent of disease. Calcific pancreatitis is not a marker of alcoholic pancreatitis alone, and rarely indicates autoimmune pancreatitis. Endoscopic ultrasound provides a better assessment of the disease than computed tomography and is useful to disclose indolent or unsuspected cancer, which can occur in up to 10% of patients.
The nidus of inflammation in chronic pancreatitis due to any cause is the head of the gland. Therefore, treatment approaches that address the disease in the head have the best long-term results. The Whipple procedure, the Beger procedure, and the Frey procedure, with or without longitudinal duct drainage, are the best surgical options, as all three approaches remove all or most of the disease in the head of the gland.
The precursor lesion that probably leads to most cases of ductular adenocarcinoma is the ductal epithelial hyperplasia/dysplasia process described by the pancreatic intraepithelial neoplasia classification system. Pancreatic intraepithelial neoplasia 2 and pancreatic intraepithelial neoplasia 3 lesions may be associated with other, nonspecific changes in pancreatic morphology seen on imaging studies, or may only be seen histologically. Resection margins for pancreatic neoplasms should be examined for advanced pancreatic intraepithelial neoplasia stage patterns of ductal hyperplasia to ensure adequate resection status.
Intraductal papillary mucinous neoplasms are small macroscopic polypoid or plaque-like adenomas that develop in the main pancreatic duct or in side-branch ducts, and secrete mucin. They are often silent symptomatically, but cause characteristic appearances of small cyst-like collections of mucus, or diffuse dilatation of the main pancreatic duct with mucus. These premalignant lesions may be multifocal or single and can evolve into invasive adenocarcinoma in a similar pattern as with other adenomatous polypoid lesions of the gastrointestinal tract. They have been diagnosed with increasing frequency, and account for more than one third of pancreatic resections at some centers. Main-duct intraductal papillary mucinous neoplasms are an indication for resection; side-branch intraductal papillary mucinous neoplasms have a lower incidence of malignancy and are sometimes followed with serial imaging surveillance.
ANATOMY
The pancreas is perhaps the most unforgiving organ in the human body, leading most surgeons to avoid even palpating it unless necessary. Situated deep in the center of the abdomen, the pancreas is surrounded by numerous important structures and major blood vessels. Seemingly minor trauma to the pancreas can result in the release of pancreatic enzymes and cause life-threatening pancreatitis. Therefore, knowledge of the relationships of the pancreas to surrounding structures is critically important for all surgeons to ensure that pancreatic injury is avoided during abdominal surgery.
The pancreas is a retroperitoneal organ that lies in an oblique position, sloping upward from the C-loop of the duodenum to the splenic hilum (Fig. 33-1). In an adult, the pancreas weighs 75 to 100 g and is about 15 to 20 cm long. The fact that the pancreas is situated so deeply in the abdomen and is sealed in the retroperitoneum explains the poorly localized and sometimes ill-defined nature with which pancreatic pathology presents. Patients with pancreatic cancer without bile duct obstruction usually present after months of vague upper abdominal discomfort, or no antecedent symptoms at all. Due to its retroperitoneal location, pain associated with pancreatitis often is characterized as penetrating through to the back.
Figure 33-1.
Pancreatic anatomy as seen on computed tomography. Knowledge of the relationship of the pancreas with surrounding structures is important to ensure that injury is avoided during abdominal surgery. IMV = inferior mesenteric vein; SMA = superior mesenteric artery; SMV = superior mesenteric vein.
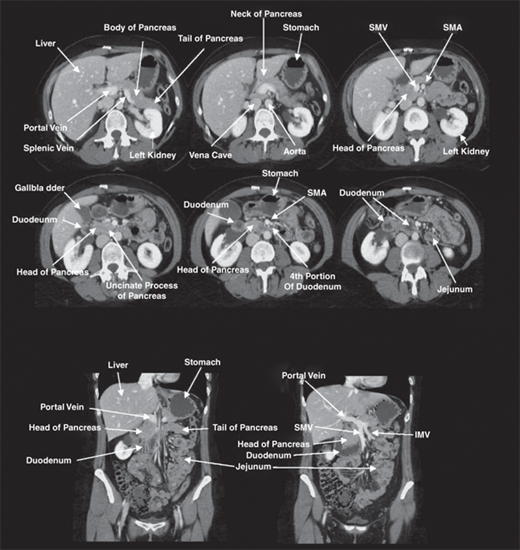
Surgeons typically describe the location of pathology within the pancreas in relation to four regions: the head, neck, body, and tail. The head of the pancreas is nestled in the C-loop of the duodenum and is posterior to the transverse mesocolon. Just posterior to the head of the pancreas lie the vena cava, the right renal artery, and both renal veins. The neck of the pancreas lies directly anterior to the portal vein. At the inferior border of the neck of the pancreas, the superior mesenteric vein joins the splenic vein and then continues toward the porta hepatis as the portal vein. The inferior mesenteric vein often joins the splenic vein near its junction with the portal vein. Sometimes, the inferior mesenteric vein joins the superior mesenteric vein or merges with the superior mesenteric portal venous junction to form a trifurcation (Fig. 33-2). The superior mesenteric artery lies parallel to and just to the left of the superior mesenteric vein. The uncinate process and the head of the pancreas wrap around the right side of the portal vein and end posteriorly near the space between the superior mesenteric vein and superior mesenteric artery. Venous branches draining the pancreatic head and uncinate process enter along the right lateral and posterior sides of the portal vein. There are usually no anterior venous tributaries, and a plane can usually be developed between the neck of the pancreas and the portal and superior mesenteric veins during pancreatic resection, unless the tumor is invading the vein anteriorly. The common bile duct runs in a deep groove on the posterior aspect of the pancreatic head until it passes through the pancreatic parenchyma to join the main pancreatic duct at the ampulla of Vater. The body and tail of the pancreas lie just anterior to the splenic artery and vein. The vein runs in a groove on the back of the pancreas and is fed by multiple fragile venous branches from the pancreatic parenchyma. These branches must be divided to perform a spleen-sparing distal pancreatectomy. The splenic artery runs parallel and just superior to the vein along the posterior superior edge of the body and tail of the pancreas. The splenic artery often is tortuous. The anterior surface of the body of the pancreas is covered by peritoneum. Once the gastrocolic omentum is divided, the body and tail of the pancreas can be seen along the floor of the lesser sac, just posterior to the stomach.
Figure 33-2.
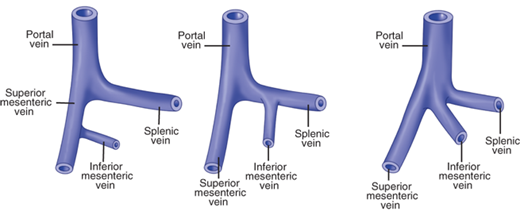
Variations in portal venous anatomy. The superior mesenteric vein joins the splenic vein and then continues toward the porta hepatis as the portal vein. The inferior mesenteric vein often joins the splenic vein near its junction with the portal vein, but sometimes joins the superior mesenteric vein; or the three veins merge as a trifurcation to form the portal vein.
Pancreatic pseudocysts commonly develop in this area, and the posterior aspect of the stomach can form the anterior wall of the pseudocyst, allowing drainage into the stomach. The base of the transverse mesocolon attaches to the inferior margin of the body and tail of the pancreas. The transverse mesocolon often forms the inferior wall of pancreatic pseudocysts or inflammatory processes, allowing surgical drainage through the transverse mesocolon. The body of the pancreas is anterior to the aorta at the origin of the superior mesenteric artery. The neck of the pancreas is anterior to the vertebral body of L1 and L2, and blunt anteroposterior trauma can compress the neck of the pancreas against the spine, causing parenchymal and, sometimes, ductal injury. The neck divides the pancreas into approximately two equal halves. The small portion of the pancreas anterior to the left kidney is referred to as the tail and is nestled in the hilum of the spleen near the splenic flexure of the left colon. Awareness of these anatomic relationships is important to avoid injury to the pancreatic tail during left colectomy or splenectomy.
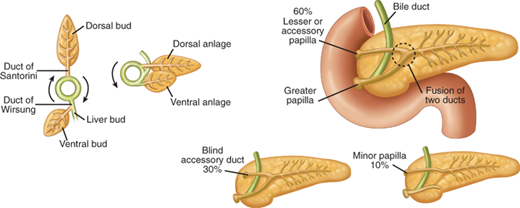
An understanding of embryology is required to appreciate the common variations in pancreatic duct anatomy. The pancreas is formed by the fusion of a ventral and dorsal bud (Fig. 33-3). The duct from the smaller ventral bud, which arises from the hepatic diverticulum, connects directly to the common bile duct. The duct from the larger dorsal bud, which arises from the duodenum, drains directly into the duodenum. The duct of the ventral anlage becomes the duct of Wirsung, and the duct from the dorsal anlage becomes the duct of Santorini. With gut rotation, the ventral anlage rotates to the right and around the posterior side of the duodenum to fuse with the dorsal bud. The ventral anlage becomes the inferior portion of the pancreatic head and the uncinate process, while the dorsal anlage becomes the body and tail of the pancreas. The ducts from each anlage usually fuse together in the pancreatic head such that most of the pancreas drains through the duct of Wirsung, or main pancreatic duct, into the common channel formed from the bile duct and pancreatic duct. The length of the common channel is variable. In about one third of patients, the bile duct and pancreatic duct remain distinct to the end of the papilla, the two ducts merge at the end of the papilla in another one third, and in the remaining one third, a true common channel is present for a distance of several millimeters. Commonly, the duct from the dorsal anlage, the duct of Santorini, persists as the lesser pancreatic duct, and sometimes drains directly into the duodenum through the lesser papilla just proximal to the major papilla. In approximately 30% of patients, the duct of Santorini ends as a blind accessory duct and does not empty into the duodenum. In 10% of patients, the ducts of Wirsung and Santorini fail to fuse.1 This results in the majority of the pancreas draining through the duct of Santorini and the lesser papilla, while the inferior portion of the pancreatic head and uncinate process drains through the duct of Wirsung and major papilla. This normal anatomic variant, which occurs in one out of 10 patients, is referred to as pancreas divisum (Fig. 33-3). In a minority of these patients, the minor papilla can be inadequate to handle the flow of pancreatic juices from the majority of the gland. This relative outflow obstruction can result in pancreatitis and is sometimes treated by sphincteroplasty of the minor papilla.
Figure 33-3.
Embryology of pancreas and duct variations. The duct of Wirsung from the ventral bud connects to the bile duct, while the duct of Santorini from the larger dorsal bud connects to the duodenum. With gut rotation, the two ducts fuse in most cases such that the majority of the pancreas drains through the duct of Wirsung to the major papilla. The duct of Santorini can persist as a blind accessory duct or drain through the lesser papilla. In a minority of patients, the ducts remain separate, and the majority of the pancreas drains through the duct of Santorini, a condition referred to as pancreas divisum.
The main pancreatic duct is usually only 2 to 3 mm in diameter and runs midway between the superior and inferior borders of the pancreas, usually closer to the posterior than to the anterior surface. Pressure inside the pancreatic duct is about twice that in the common bile duct, which is thought to prevent reflux of bile into the pancreatic duct. The main pancreatic duct joins with the common bile duct and empties at the ampulla of Vater or major papilla, which is located on the medial aspect of the second portion of the duodenum. The muscle fibers around the ampulla form the sphincter of Oddi, which controls the flow of pancreatic and biliary secretions into the duodenum. Contraction and relaxation of the sphincter is regulated by complex neural and hormonal factors. When the accessory pancreatic duct or lesser duct drains into the duodenum, a lesser papilla can be identified approximately 2 cm proximal to the ampulla of Vater.
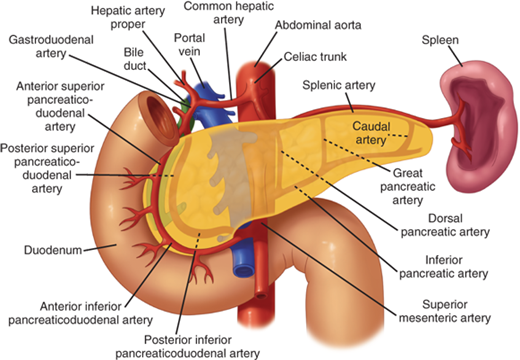
The blood supply to the pancreas comes from multiple branches from the celiac and superior mesenteric arteries (Fig. 33-4). The common hepatic artery gives rise to the gastroduodenal artery before continuing toward the porta hepatis as the proper hepatic artery. The right gastric artery branches off the gastroduodenal artery just superior to the duodenum. The gastroduodenal artery then travels inferiorly anterior to the neck of the pancreas and posterior to the duodenal bulb. A posterior ulcer in the duodenal bulb can erode into the gastroduodenal artery in this location. At the inferior border of the duodenum, the gastroduodenal artery then gives rise to the right gastroepiploic artery then continues on as the anterior superior pancreaticoduodenal artery, which branches into the anterior and posterior superior pancreaticoduodenal arteries. As the superior mesenteric artery passes behind the neck of the pancreas, it gives off the inferior pancreaticoduodenal artery at the inferior margin of the neck of the pancreas. This vessel quickly divides into the anterior and posterior inferior pancreaticoduodenal arteries. The superior and inferior pancreaticoduodenal arteries join together within the parenchyma of the anterior and posterior sides of the head of the pancreas along the medial aspect of the C-loop of the duodenum to form arcades that give off numerous branches to the duodenum and head of the pancreas. Therefore, it is impossible to resect the head of the pancreas without devascularizing the duodenum, unless a rim of pancreas containing the pancreaticoduodenal arcade is preserved. Variations in the arterial anatomy occur in one out of five patients. The right hepatic artery, common hepatic artery, or gastroduodenal arteries can arise from the superior mesenteric artery. In 15% to 20% of patients, the right hepatic artery will arise from the superior mesenteric artery and travel upwards toward the liver along the posterior aspect of the head of the pancreas (referred to as a replaced right hepatic artery). It is important to look for this variation on preoperative computed tomographic (CT) scans and in the operating room so the replaced hepatic artery is recognized and injury is avoided. The body and tail of the pancreas are supplied by multiple branches of the splenic artery. The splenic artery arises from the celiac trunk and travels along the posterior-superior border of the body and tail of the pancreas toward the spleen. The inferior pancreatic artery usually arises from the superior mesenteric artery and runs to the left along the inferior border of the body and tail of the pancreas, parallel to the splenic artery. Three vessels run perpendicular to the long axis of the pancreatic body and tail and connect the splenic artery and inferior pancreatic artery. They are, from medial to lateral, the dorsal, great, and caudal pancreatic arteries. These arteries form arcades within the body and tail of the pancreas, and account for the rich blood supply of the organ.
The venous drainage of the pancreas follows a pattern similar to that of the arterial supply (Fig. 33-5). The veins are usually superficial to the arteries within the parenchyma of the pancreas. There is an anterior and posterior venous arcade within the head of the pancreas. The superior veins drain directly into the portal vein just above the neck of the pancreas. The posterior inferior arcade drains directly into the inferior mesenteric vein at the inferior border of the neck of the pancreas. These venous tributaries must be divided during a Whipple procedure. The anterior inferior pancreaticoduodenal vein joins the right gastroepiploic vein and the middle colic vein to form a common venous trunk, which enters into the superior mesenteric vein. Traction on the transverse colon during colectomy can tear these fragile veins, which then retract into the parenchyma of the pancreas, making control tedious. There also are numerous small venous branches coming from the pancreatic parenchyma directly into the lateral and posterior aspect of the portal vein. Venous return from the body and tail of the pancreas drains into the splenic vein.
Figure 33-5.
Venous drainage from the pancreas. The venous drainage of the pancreas follows a pattern similar to the arterial supply, with the veins usually superficial to the arteries. Anterior traction on the transverse colon can tear fragile branches along the inferior border of the pancreas, which then retract into the parenchyma of the pancreas. Venous branches draining the pancreatic head and uncinate process enter along the right lateral and posterior sides of the portal vein. There are usually no anterior venous tributaries, and a plane can usually be developed between the neck of the pancreas and the portal and superior mesenteric veins.
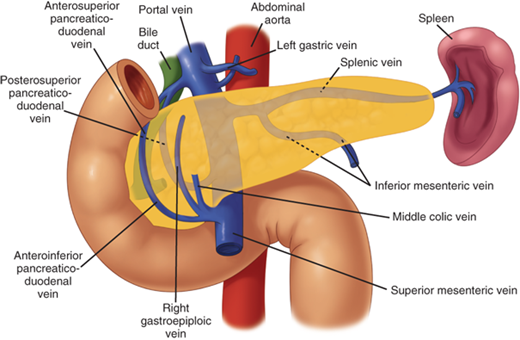
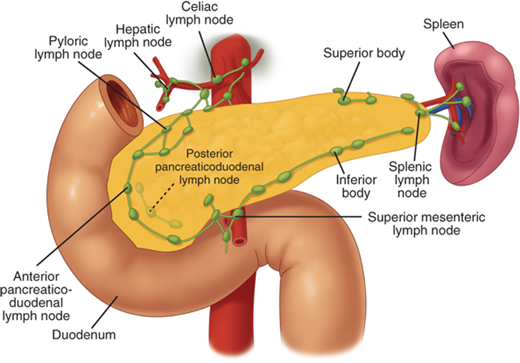
The lymphatic drainage from the pancreas is diffuse and widespread (Fig. 33-6). The profuse network of lymphatic vessels and lymph nodes draining the pancreas provides egress to tumor cells arising from the pancreas. This diffuse lymphatic drainage contributes to the fact that pancreatic cancer often presents with positive lymph nodes and a high incidence of local recurrence after resection. Lymph nodes can be palpated along the distal bile duct and posterior aspect of the head of the pancreas in the pancreaticoduodenal groove, where the mesenteric vein passes under the neck of the pancreas, along the inferior border of the body, at the celiac axis and along the hepatic artery ascending into the porta hepatis, and along the splenic artery and vein. The pancreatic lymphatics also communicate with lymph nodes in the transverse mesocolon and mesentery of the proximal jejunum. Tumors in the body and tail of the pancreas often metastasize to these nodes and lymph nodes along the splenic vein and in the hilum of the spleen.
Figure 33-6.
Lymphatic supply to the pancreas. The lymphatic drainage from the pancreas is diffuse and widespread, which explains the high incidence of lymph node metastases and local recurrence of pancreatic cancer. The pancreatic lymphatics also communicate with lymph nodes in the transverse mesocolon and mesentery of the proximal jejunum. Tumors in the body and tail of the pancreas are often unresectable because they metastasize to these lymph nodes. (Reproduced with permission from Bell RH Jr: Atlas of pancreatic surgery, in Bell RH Jr, Rikkers LF, Mulholland MW (eds): Digestive Tract Surgery: A Text and Atlas. Philadelphia: Lippincott-Raven, 1996, p 969.)
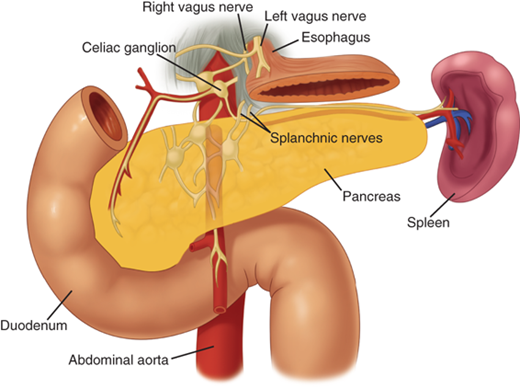
The pancreas is innervated by the sympathetic and parasympathetic nervous systems. The acinar cells responsible for exocrine secretion, the islet cells responsible for endocrine secretion, and the islet vasculature are innervated by both systems. The parasympathetic system stimulates endocrine and exocrine secretion and the sympathetic system inhibits secretion.2The pancreas is also innervated by neurons that secrete amines and peptides, such as somatostatin, vasoactive intestinal peptide (VIP), calcitonin gene-related peptide (CGRP), and galanin. The exact role of these neurons in pancreatic physiology is uncertain, but they do appear to affect both exocrine and endocrine function. The pancreas also has a rich supply of afferent sensory fibers, which are responsible for the intense pain associated with advanced pancreatic cancer, as well as acute and chronic pancreatitis. These somatic fibers travel superiorly to the celiac ganglia (Fig. 33-7). Interruption of these somatic fibers can stop transmission of pain sensation.
Figure 33-7.
Innervation of the pancreas. The pancreas has a rich supply of afferent sensory fibers that travel superiorly to the celiac ganglia. Interruption of these somatic fibers with a celiac plexus block can interfere with transmission of pancreatic pain. (Reproduced with permission from Bell RH Jr: Atlas of pancreatic surgery, in Bell RH Jr, Rikkers LF, Mulholland MW [eds]: Digestive Tract Surgery: A Text and Atlas. Philadelphia: Lippincott-Raven, 1996, p 969.)
HISTOLOGY AND PHYSIOLOGY
The exocrine pancreas accounts for about 85% of the pancreatic mass; 10% of the gland is accounted for by extracellular matrix, and 4% by blood vessels and the major ducts, whereas only 2% of the gland is comprised of endocrine tissue. The endocrine and exocrine pancreas are sometimes thought of as functionally separate, but these different components of the organ are coordinated to allow an elegant regulatory feedback system for digestive enzyme and hormone secretion. This complex system regulates the type of digestion, its rate, and the processing and distribution of absorbed nutrients. This coordination is facilitated by the physical approximation of the islets and the exocrine pancreas, the presence of specific islet hormone receptors on the plasma membranes of pancreatic acinar cells, and the existence of an islet-acinar portal blood system.
Although patients can live without a pancreas when insulin and digestive enzyme replacement are administered, the loss of this islet-acinar coordination leads to impairments in digestive function. Although only approximately 20% of the normal pancreas is required to prevent insufficiency, in many patients undergoing pancreatic resection, the remaining pancreas is not normal, and pancreatic endocrine and exocrine insufficiency can develop with removal of smaller portions of the gland.
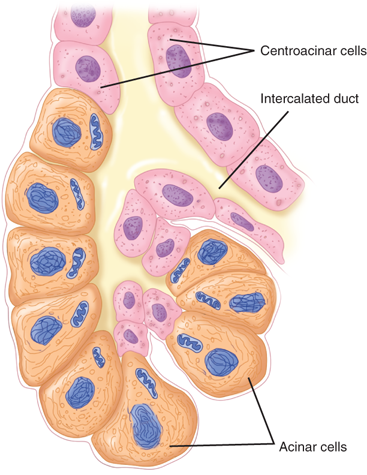
The pancreas secretes approximately 500 to 800 mL per day of colorless, odorless, alkaline, isosmotic pancreatic juice. Pancreatic juice is a combination of acinar cell and duct cell secretions. The acinar cells secrete amylase, proteases, and lipases, enzymes responsible for the digestion of all three food types: carbohydrate, protein, and fat. The acinar cells are pyramid-shaped, with their apices facing the lumen of the acinus. Near the apex of each cell are numerous enzyme-containing zymogen granules that fuse with the apical cell membrane (Fig. 33-8). Unlike the endocrine pancreas, where islet cells specialize in the secretion of one hormone type, individual acinar cells secrete all types of enzymes. However, the ratio of the different enzymes released is adjusted to the composition of digested food through nonparallel regulation of secretion.
Pancreatic amylase is secreted in its active form and completes the digestive process already begun by salivary amylase. Amylase is the only pancreatic enzyme secreted in its active form, and it hydrolyzes starch and glycogen to glucose, maltose, maltotriose, and dextrins. These simple sugars are transported across the brush border of the intestinal epithelial cells by active transport mechanisms. Gastric hydrolysis of protein yields peptides that enter the intestine and stimulate intestinal endocrine cells to release cholecystokinin (CCK)-releasing peptide, CCK, and secretin, which then stimulate the pancreas to secrete enzymes and bicarbonate into the intestine.
The proteolytic enzymes are secreted as proenzymes that require activation. Trypsinogen is converted to its active form, trypsin, by another enzyme, enterokinase, which is produced by the duodenal mucosal cells. Trypsin, in turn, activates the other proteolytic enzymes. Trypsinogen activation within the pancreas is prevented by the presence of inhibitors that are also secreted by the acinar cells. A failure to express a normal trypsinogen inhibitor, pancreatic secretory trypsin inhibitor (PSTI), also known as serine protease inhibitor Kazal type 1 (SPINK1), is a cause of familial pancreatitis. Inhibition of trypsinogen activation ensures that the enzymes within the pancreas remain in an inactive precursor state and are activated only within the duodenum. Trypsinogen is expressed in several isoforms, and a missense mutation on the cationic trypsinogen, or PRSS1, results in premature, intrapancreatic activation of trypsinogen. This accounts for about two-thirds of cases of hereditary pancreatitis. Chymotrypsinogen is activated to form chymotrypsin. Elastase, carboxypeptidase A and B, and phospholipase are also activated by trypsin. Trypsin, chymotrypsin, and elastase cleave bonds between amino acids within a target peptide chain, and carboxypeptidase A and B cleave amino acids at the end of peptide chains. Individual amino acids and small dipeptides are then actively transported into the intestinal epithelial cells. Pancreatic lipase hydrolyzes triglycerides to 2-monoglyceride and fatty acid. Pancreatic lipase is secreted in an active form. Colipase is also secreted by the pancreas and binds to lipase, changing its molecular configuration and increasing its activity. Phospholipase A2 is secreted by the pancreas as a proenzyme that becomes activated by trypsin. Phospholipase A2 hydrolyzes phospholipids and, as with all lipases, requires bile salts for its action. Carboxylic ester hydrolase and cholesterol esterase hydrolyze neutral lipid substrates like esters of cholesterol, fat-soluble vitamins, and triglycerides. The hydrolyzed fat is then packaged into micelles for transport into the intestinal epithelial cells, where the fatty acids are reassembled and packaged inside chylomicrons for transport through the lymphatic system into the bloodstream (Table 33-1).
| ENZYME | SUBSTRATE | PRODUCT |
|---|---|---|
Carbohydrate Amylase (active) | Starch, glycogen | Glucose, maltose, maltotriose, dextrins |
Protein Endopeptidases Trypsinogen (inactive) Chymotrypsinogen (inactive) Proelastase (inactive) Exopeptidases Procarboxy peptidase A&B (inactive) A&B (active) | Cleave bonds between amino acids Cleave amino acids from end of peptide chains | Amino acids, dipeptides — |
Fat Pancreatic lipase (active) Phospholipase A2 (inactive) Cholesterol esterase | Triglycerides Phospholipase Neutral lipids | 2-Monoglycerides, fatty acids — — |





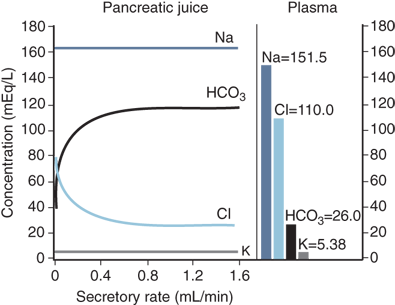
The centroacinar and intercalated duct cells secrete the water and electrolytes present in the pancreatic juice. About 40 acinar cells are arranged into a spherical unit called an acinus. Centroacinar cells are located near the center of the acinus and are responsible for fluid and electrolyte secretion. These cells contain the enzyme carbonic anhydrase, which is needed for bicarbonate secretion. The amount of bicarbonate secreted varies with the pancreatic secretory rate, with greater concentrations of bicarbonate being secreted as the pancreatic secretory rate increases. Chloride secretion varies inversely with bicarbonate secretion such that the sum of these two remains constant. In contrast, sodium and potassium concentrations are kept constant throughout the spectrum of secretory rates3 (Fig. 33-9). The hormone secretin is released from cells in the duodenal mucosa in response to acidic chyme passing through the pylorus into the duodenum. Secretin is the major stimulant for bicarbonate secretion, which buffers the acidic fluid entering the duodenum from the stomach. CCK also stimulates bicarbonate secretion, but to a much lesser extent than secretin. CCK potentiates secretin-stimulated bicarbonate secretion. Gastrin and acetylcholine, both stimulants of gastric acid secretion, are also weak stimulants of pancreatic bicarbonate secretion.4 Truncal vagotomy produces a myriad of complex effects on the downstream digestive tract, but the sum effect on the exocrine pancreas is a reduction in bicarbonate and fluid secretion.5 The endocrine pancreas also influences the adjacent exocrine pancreatic secretions. Somatostatin, pancreatic polypeptide (PP), and glucagon are all thought to inhibit exocrine secretion.
Figure 33-9.
Composition of pancreatic exocrine secretions. Greater concentrations of bicarbonate are secreted at higher secretory rates, and chloride secretion varies inversely with bicarbonate secretion. In contrast, sodium and potassium concentrations are independent of the secretory rate. (Reproduced with permission from Bro-Rasmussen F et al: The composition of pancreatic juice as compared to sweat, parotid saliva, and tears. Acta Phys Scandinav. 37:97–113, 1956.)
The acinar cells release pancreatic enzymes from their zymogen granules into the lumen of the acinus, and these proteins combine with the water and bicarbonate secretions of the centroacinar cells. The pancreatic juice then travels into small intercalated ducts. Several small intercalated ducts join to form an interlobular duct. Cells in the interlobular ducts contribute fluid and electrolytes to adjust the final concentrations of the pancreatic fluid. Interlobular ducts then join to form about 20 secondary ducts that empty into the main pancreatic duct. Destruction of the branching ductal tree from recurrent inflammation, scarring, and deposition of stones eventually contributes to destruction of the exocrine pancreas and exocrine pancreatic insufficiency.
There are nearly 1 million islets of Langerhans in the normal adult pancreas. They vary greatly in size from 40 to 900 μm. Larger islets are located closer to the major arterioles and smaller islets are embedded more deeply in the parenchyma of the pancreas. Most islets contain 3000 to 4000 cells of five major types: alpha cells that secrete glucagon, β-cells that secrete insulin, delta cells that secrete somatostatin, epsilon cells that secrete ghrelin, and PP cells that secrete PP (Table 33-2).
| HORMONES | ISLET CELL | FUNCTIONS |
|---|---|---|
| Insulin | β (beta cell) | Decreased gluconeogenesis, glycogenolysis, fatty acid breakdown, and ketogenesis Increased glycogenesis, protein synthesis, and glucose uptake |
| Glucagon | α (alpha cell) | Opposite effects of insulin; increased hepatic glycogenolysis and gluconeogenesis |
| Somatostatin | δ (delta cell) | Inhibits GI secretion Inhibits secretion and action of all GI endocrine peptides Inhibits cell growth |
| Pancreatic polypeptide | PP (PP cell) | Inhibits pancreatic exocrine secretion and section of insulin Facilitates hepatic effect of insulin |
| Amylin (IAPP) | β (beta cell) | Counterregulates insulin secretion and function |
| Pancreastatin | β (beta cell) | Decreases insulin and somatostatin release Increases glucagon release Decreases pancreatic exocrine secretion |
| Ghrelin | (epsilon cell) | Decreases insulin release and insulin action |
Insulin is the best-studied pancreatic hormone. The discovery of insulin in 1920 by Frederick Banting, an orthopedic surgeon, and Charles Best, a medical student, was recognized with the awarding of the Nobel Prize in Physiology or Medicine. They produced diabetes in dogs by performing total pancreatectomy and then treated them with crude pancreatic extracts from dog and calf pancreata using techniques to prevent the breakdown of insulin by the proteolytic enzymes of the exocrine pancreas. Insulin was subsequently purified and found to be a 56-amino acid peptide with two chains, an α and a β chain, joined by two disulfide bridges and a connecting peptide, or C-peptide. Proinsulin is made in the endoplasmic reticulum and then is transported to the Golgi complex, where it is packaged into granules and the C-peptide is cleaved off. There are two phases of insulin secretion. In the first phase, stored insulin is released. This phase lasts about 5 minutes after a glucose challenge. The second phase of insulin secretion is a longer, sustained release due to ongoing production of new insulin. β-cell synthesis of insulin is regulated by plasma glucose levels, neural signals, and the paracrine influence of other islet cells. The diagnosis of diabetes is made by using oral and intravenous (IV) glucose tolerance tests. Oral glucose not only enters the bloodstream but also stimulates the release of enteric hormones such as gastric inhibitory peptide (also known as glucose-dependentinsulinotropic polypeptide or GIP), glucagon-like peptide-1 (GLP-1), and CCK that augment the secretion of insulin, and are therefore referred to as incretins. As a result, oral glucose is a more vigorous stimulus to insulin secretion than IV glucose. In the oral glucose tolerance test (OGTT), the patient is fasted overnight, and a basal glucose value is determined. Forty g/m2or 75 g of glucose is given orally over 10 minutes. Blood samples are taken every 30 minutes for 2 hours. Normal values and criteria for diabetes vary by age, but essentially all values should be <200 mg/dL, and the 120-minute value should be <140 mg/dL.
Insulin secretion by the β-cell is also influenced by plasma levels of amino acids such as arginine, lysine, leucine, and free fatty acids. Glucagon, GIP, GLP-1, and CCK stimulate insulin release, while somatostatin, amylin, and pancreastatin inhibit insulin release.6 Cholinergic fibers and beta sympathetic fibers stimulate insulin release, while alpha sympathetic fibers inhibit insulin secretion.
Insulin’s glucoregulatory function is to inhibit endogenous (hepatic) glucose production and to facilitate glucose transport into cells, thus lowering plasma glucose levels. Insulin also inhibits glycogenolysis, fatty acid breakdown, and ketone formation, and stimulates protein synthesis. There is a considerable amount of functional reserve in insulin secretory capacity. If the remaining portion of the pancreas is healthy, about 80% of the pancreas can be resected without the patient becoming diabetic.7 In patients with chronic pancreatitis, or other conditions in which much of the gland is diseased, resection of a smaller fraction of the pancreas can result in pancreatogenic, or type 3c diabetes (Table 33-3).
| PARAMETER | TYPE 1 | TYPE 2 | TYPE 3C |
|---|---|---|---|
| IDDM | NIDDM | Pancreatogenic | |
| Ketoacidosis | Common | Rare | Rare |
| Hyperglycemia | Severe | Usually Mild | Mild |
| Hypoglycemia | Common | Rare | Common |
| Peripheral Insulin Sensitivity | Normal or Increased | Decreased | Increased |
| Hepatic Insulin Sensitivity | Normal | Normal or Decreased | Decreased |
| Insulin Levels | Low | High | Low |
| Glucagon Levels | Normal or High | Normal or High | Low |
| PP Levels | Normal or Low (late) | High | Low |
| GIP Levels | Normal or Low | Normal or High | Low |
| GLP-1 Levels | Normal | Normal or High | Normal or High |
| Typical Age of Onset | Childhood or adolescence | Adulthood | Any |
Insulin receptors are dimeric, tyrosine kinase–containing transmembrane proteins that are located on all cells. Insulin deficiency (seen in type 1 and type 3c diabetes) results in an overexpression or upregulation of insulin receptors, which causes an enhanced sensitivity to insulin in muscle and adipocytes (and therefore increases the risk of insulin-induced hypoglycemia). Type 2 diabetes is associated with a downregulation of insulin receptors and relative hyperinsulinemia, with resulting insulin resistance. Some forms of diabetes are associated with selected impairments of hepatic or peripheral insulin receptors, such as pancreatogenic or type 3c diabetes (T3cDM) or maturity-onset diabetes of the young (MODY).
Glucagon is a 29-amino-acid, single-chain peptide that promotes hepatic glycogenolysis and gluconeogenesis and counteracts the effects of insulin through its hyperglycemic action. Glucose is the primary regulator of glucagon secretion, as it is with insulin, but it has an inhibitory rather than stimulatory effect. Glucagon release is stimulated by hypoglycemia, and by the amino acids arginine and alanine. GLP-1 inhibits glucagon secretion in vivo, and insulin and somatostatin inhibit glucagon secretion in a paracrine fashion within the islet. The same neural impulses that regulate insulin secretion also regulate glucagon secretion, so that the two hormones work together in a balance of actions to maintain glucose levels. Cholinergic and beta sympathetic fibers stimulate glucagon release, while alpha sympathetic fibers inhibit glucagon release.8 In pancreatogenic or type 3c diabetes, glucagon responsiveness to a fall in blood glucose is lost, thereby increasing the risk for hypoglycemia.
Although originally isolated from the hypothalamus, somatostatin is a peptide that is now known to have a wide anatomic distribution, not only in neurons but also in the pancreas, gut, and other tissues. It is a highly conserved peptide hormone, as it is found in lower vertebrates, and is now realized to be of fundamental importance in regulatory processes throughout the body. One gene encodes for a common precursor that is differentially processed to generate tissue-specific amounts of two bioactive products, somatostatin-14, and somatostatin-28. These peptides inhibit endocrine and exocrine secretion and affect neurotransmission, GI and biliary motility, intestinal absorption, vascular tone, and cell proliferation.
Five different somatostatin receptors (SSTRs) have been cloned and the biologic properties of each are being unraveled.9 All five are G-protein-coupled receptors with seven highly conserved transmembrane domains and unique amino and carboxy termini. Phosphorylation sites located within the second and third intracellular loops and in the cytoplasmic C-terminal segment are thought to mediate receptor regulation. Although the naturally occurring peptides bind to all five receptors, somatostatin-28 is relatively selective for SSTR5. The hexapeptide and octapeptide analogues such as octreotide bind only to SSTR2, SSTR3, and SSTR5. These analogues have a longer serum half-life, and their potent inhibitory effect has been used clinically to treat both endocrine and exocrine disorders. For example, octreotide has been shown to decrease fistula output and speed the time it takes for enteric and pancreatic fistulas to close.10
Endocrine release of somatostatin occurs during a meal. The major stimulant is probably intraluminal fat. Acidification of the gastric and duodenal mucosa also releases somatostatin in isolated perfused organ preparations. Acetylcholine from the cholinergic neurons inhibits somatostatin release.
Pancreatic polypeptide (PP) is a 36-amino-acid, straight-chain peptide discovered by Kimmel in 1968 during the process of insulin purification. Protein is the most potent enteral stimulator of PP release, closely followed by fat, whereas glucose has a weaker effect.11 Hypoglycemia, whether or not it is insulin induced, strongly stimulates PP secretion through cholinergic stimulation.12 Phenylalanine, tryptophan, and fatty acids in the duodenum stimulate PP release, probably by inducing CCK and secretin release. In the isolated perfused pancreas, insulin and gastric inhibitory peptide stimulate PP release, while glucagon and somatostatin inhibit it. Vagal stimulation of the pancreas is the most important regulator of PP secretion. In fact, vagotomy eliminates the rise in PP levels usually seen after a meal. This can be used as a test for the completeness of a surgical vagotomy or for the presence of diabetic autonomic neuropathy.
PP has been shown to inhibit choleresis (bile secretion), gallbladder contraction, and secretion by the exocrine pancreas. However, PP’s most important role is in glucose regulation through its regulation of hepatic insulin receptor gene expression. A deficiency in PP secretion due to proximal pancreatectomy, severe chronic pancreatitis, or cystic fibrosis,is associated with diminished hepatic insulin sensitivity due to reduced hepatic insulin receptor availability.13 This effect is reversed by PP administration.14
Recent studies have shown that a fifth islet peptide, ghrelin, is secreted from a distinct population of islet cells, called epsilon cells.15,16 Ghrelin also is present in the gastric fundus in large amounts and stimulates growth hormone secretion via growth hormone releasing hormone release from the pituitary. It is an orexigenic, or appetite-stimulating, peptide the plasma levels of which are increased in obesity. Ghrelin has also been shown to block insulin effects on the liver, and inhibits the β-cell response to incretin hormones and glucose.17 Therefore, ghrelin secretion from and within the islet may modulate the responses of other islet cells to nutrient and hormonal stimuli.
In addition to the five main peptides secreted by the pancreas, there are a number of other peptide products of the islet cells, including amylin and pancreastatin, as well as neuropeptides such as VIP, galanin, and serotonin. Amylin or islet amyloid polypeptide (IAPP) is a 37-amino-acid polypeptide that is predominantly expressed by the pancreatic β-cells, where it is stored along with insulin in secretory granules.18 The function of IAPP seems to be the modulation or counterregulation of insulin secretion and function. Pancreastatin is a recently discovered pancreatic islet peptide product that inhibits insulin, and possibly somatostatin release, and augments glucagon release.19,20 In addition to this effect on the endocrine pancreas, pancreastatin inhibits pancreatic exocrine secretion.21
The β-cells are generally located in the central portion of each islet and make up about 70% of the total islet cell mass. The other cell types are located predominantly in the periphery. The delta cells are least plentiful, making up only 5%; the α-cells make up 10%, and the PP cells make up 15%.22 In contrast to the acinar cells that secrete the full gamut of exocrine enzymes, the islet cells seem to specialize in the secretion of predominantly one hormone. However, individual islet cells can secrete multiple hormones. For example, the β-cells secrete both insulin and amylin, which counterregulates the actions of insulin. In reality, more than 20 different hormones are secreted by the islets, and the exact functions of this milieu are very complex. There is diversity among the islets depending on their location within the pancreas. The α– and δ-cells are evenly distributed throughout the pancreas, but islets in the head and uncinate process (ventral anlage) have a higher percentage of PP cells and fewer α-cells, whereas islets in the body and tail (dorsal anlage) contain the majority of α-cells and few PP cells. This is clinically significant because pancreatoduodenectomy removes 95% of the PP cells in the pancreas. This may partially explain the higher incidence of glucose intolerance after the Whipple procedure compared to a distal pancreatectomy with an equivalent amount of tissue resected. In addition, chronic pancreatitis, which disproportionately affects the pancreatic head, is associated with PP deficiency and pancreatogenic diabetes.23 The relative preponderance of α-cells in the body and tail of the pancreas explains the typical location of glucagonomas.
ACUTE PANCREATITIS
Acute pancreatitis is a common and challenging disease that can develop both local and systemic complications. Its hallmark is acute pancreatic inflammation associated with little or no fibrosis. It ranges from a mild self-limiting inflammation of the pancreas to critical disease characterized by infected pancreatic necrosis, multiple organ failure and a high risk of mortality.24 The clinical outcome has improved over recent decades, even in the absence of specific treatments that target outcome-determining pathophysiology, probably because of a more consistent approach to diagnosis, monitoring and management.
Acute pancreatitis is the most common gastrointestinal discharge diagnosis in the United States (274,119 patients in 2009), an incidence which has increased 30% since 2000, and is associated with the highest aggregate inpatient costs at 2.6 billion dollars per year.25 The crude mortality rate of 1.0/100,000 ranks it as the 14th most fatal illness overall and the ninth most common noncancer gastrointestinal death. Worldwide the incidence of acute pancreatitis ranges from 5 to 80/100,000 population with the highest incidence recorded in Finland and United States.26 The racial incidence of acute pancreatitis also shows significant variation related to the prevalence of etiological factors and ethnicity. The annual incidence of acute pancreatitis in Native Americans is 4 per 100,000 population; in whites it is 5.7 per 100,000 population; and in blacks it is 20.7 per 100,000 population.27 Smoking is an independent risk factor for acute pancreatitis.28
Many factors have been causally related to the onset of acute pancreatitis (Table 33-4), but in many instances the mechanism is poorly understood. The most common causes are gallstones and alcohol, accounting for up to 80% of cases, but it is not uncommon to diagnose acute pancreatitis in the absence of these etiological factors. Therefore it is important that a systematic approach is taken to the identification of other, less common, and potentially modifiable, factors. The median age of the onset of acute pancreatitis varies with etiology; alcohol and drug induced pancreatitis typically present in the third or fourth decade compared with gallstone and trauma induced disease in the sixth decade. The gender difference is probably more related to etiology: in males alcohol is more often the cause while in females it is gallstones.
Alcohol Biliary tract disease Hyperlipidemia Hereditary Hypercalcemia Trauma External Surgical Endoscopic retrograde cholangiopancreatography Ischemia Hypoperfusion Atheroembolic Vasculitis Pancreatic duct obstruction Neoplasms Pancreas divisum Ampullary and duodenal lesions Infections Venom Drugs Idiopathic |
Evidence that passage of a gallstone is related to the onset of acute pancreatitis comes from the characteristic transient derangement of liver function tests and the high retrieval rate of gallstones from feces within 10 days of an attack of acute pancreatitis compared with those without acute pancreatitis (88% vs. 11%).29 The mechanism by which small gallstones migrating down the common bile duct, past the pancreatic duct junction and into the duodenum, cause acute pancreatitis is not clear. Opie made the seminal observation of a gallstone impacted in the sphincter of Oddi in two fatal cases of acute pancreatitis, which lead to the “common channel” hypothesis. It was proposed that a gallstone transiently lodged in the distal common channel of the ampulla of Vater allowed bile to reflux into the pancreatic duct, but this cannot be reliably reproduced in experimental models. Another proposal suggested that transient incompetence caused by the passage of a stone through the sphincter might allow duodenal fluid and bile to reflux into the pancreatic duct, but this is refuted by the usual absence of acute pancreatitis after endoscopic sphincterotomy or surgical sphinteroplasty. A third possibility is that acute pancreatitis is due to a gallstone obstructing the pancreatic duct, leading to ductal hypertension. It has been postulated that this backpressure might lead to minor ductal disruption, extravasation of pancreatic juice into the less alkaline interstitium of the pancreas, and promotion of enzyme activation. When gallstones and other etiological factors cannot be identified there is still the possibility of finding microlithiasis, seen as birefringent crystals, on bile microscopy.30 This occult microlithiasis is probably responsible for up to half of those with idiopathic acute pancreatitis.
Alcohol ingestion is associated with acute pancreatitis and sustained alcohol ingestion is associated with recurrent acute pancreatitis and development of chronic pancreatitis in susceptible individuals who have been drinking for more than a decade. The type of alcohol consumed is less important than the amount consumed (typically 100 to 150 grams per day) and the pattern of drinking. It is common for patients with alcohol associated acute pancreatitis to have a history of excess alcohol consumption prior to the first attack. There are several mechanisms by which ethanol causes acute pancreatitis. Ethanol is a metabolic toxin to pancreatic acinar cells and causes a brief secretory increase followed by inhibition. The secretory burst coupled with ethanol induced spasm of the sphincter of Oddi is thought to incite acute pancreatitis. Ethanol also induces ductal permeability, which would allow prematurely activated enzymes to cause damage to the pancreatic parenchyma. Ethanol also increases the protein content of pancreatic juice, decreases bicarbonate levels, and trypsin inhibitor concentration. The formation of protein plugs may also contribute by causing an obstructive element to pancreatic outflow.
Acute pancreatitis can result from a number of treatments, including pancreatic biopsy, exploration of the extrahepatic biliary tree and ampulla of Vater, distal gastrectomy, splenectomy, colectomy, nephrectomy, aortic aneurysmorraphy, and retroperitoneal lymphadenectomy. As the pancreas is susceptible to ischemia it can also occur secondary to splanchnic hypoperfusion with cardio-pulmonary bypass or cardiac transplant. Most commonly, acute pancreatitis occurs as a complication of ERCP in 5% to 10% of procedures, and in many series it is the third most common identified etiological factor. The risk of post-ERCP acute pancreatitis is increased if the contrast agent is infused repeatedly under high pressure by the endoscopist and in patients with sphincter of Oddi dysfunction. Recent evidence demonstrates that the risk can be decreased with prophylactic, rectally administered, nonsteroidal drugs.31
Hereditary pancreatitis is an autosomal dominant disorder usually related to mutations of the cationic trypsinogen gene (PRSS1). Mutations in this gene cause premature activation of trypsinogen to trypsin and cause abnormalities of ductal secretion, both of which promote acute pancreatitis. Mutations in the SPINK1 protein, which blocks the active binding site of trypsin, is also likely to have a role in predisposing to acute pancreatitis. Variations in penetration and phenotype are common and there are many other mutations that may become implicated. Mutant enzymes activated within acinar cells can overwhelm the first line of defense (pancreatic secretory trypsin inhibitor) and resist backup defenses (e.g., proteolytic degradation, enzyme Y, and trypsin itself) allowing activated mutant cationic trypsin to trigger the entire zymogen activation cascade.32
A pancreatic or periampullary tumor should be considered in any patient with idiopathic acute pancreatitis. Approximately 1% to 2% of patients with acute pancreatitis have a pancreatic tumor, and an episode of acute pancreatitis can be the first clinical manifestation of the tumor. If no historical information leads to an etiologic diagnosis, cross sectional pancreatic imaging after the resolution of an unexplained episode of acute pancreatitis is indicated.
Patients with types I and V hyperlipoproteinemia can experience episodes of abdominal pain, and these often occur in association with marked hypertriglyceridaemia. Lipase is thought to liberate toxic fatty acids into the pancreatic microcirculation, leading to microcirculatory impairment and ischemia. Dietary modifications to restrict triglycerides are usually effective, but clofibrate may be prescribed in refractory cases of hypertriglyceridemia.
Many drugs can produce hyperamylasemia and/or abdominal pain, and a drug is considered suspect if the pancreatitis-like illness resolves with its discontinuation. Ethical considerations generally rule out rechallenge with the suspect drug, so the connection often remains vague. However, despite these limitations, certain drugs are known to be capable of causing acute pancreatitis. These include the thiazide diuretics, furosemide, estrogens, azathioprine, l-asparaginase, 6-mercaptopurine, methyldopa, the sulfonamides, tetracycline, pentamidine, procainamide, nitrofurantoin, dideoxyinosine, valproic acid, and acetylcholinesterase inhibitors. In addition, lipid-based intravenous drugs and solutions, such as propofol, can also cause acute pancreatitis.33 A history of verified or suspected drug-induced pancreatitis should serve as a contraindication to prescribing that medication again.
Hypercalcemic states arising from hyperparathyroidism can result in both acute and chronic pancreatitis; the mechanism most likely involves hypersecretion and the formation of calcified stones intraductally. Also implicated are infestations by Ascaris lumbricoides and the liver fluke Clonorchis sinensis, which is endemic to China, Japan, and Southeast Asia. These cause Oriental cholangitis, which is associated with cholangiocarcinoma obstructing the pancreatic duct.
Other implicated factors include azotemia, vasculitis, and the sting of the Trinidadian scorpion Tityus trinitatis. This scorpion’s venom has been shown to cause neurotransmitter discharge from cholinergic nerve terminals, leading to massive production of pancreatic juice. Poisoning with antiacetylcholinesterase insecticides has a similar effect. Finally, no apparent cause can be ascribed to some episodes of acute pancreatitis, and these constitute the group referred to as idiopathic pancreatitis. Some of these patients are eventually found to have gallstone-related pancreatitis, which calls for caution in labeling any episode “idiopathic.”
Acute pancreatitis occurs in various degrees of severity, the determinants of which are multifactorial. The generally prevalent belief today is that pancreatitis begins with the activation of digestive zymogens inside acinar cells, which cause acinar cell injury. Studies suggest that the ultimate severity of the resulting pancreatitis may be determined by the events that occur subsequent to acinar cell injury.34 These include inflammatory cell recruitment and activation, as well as generation and release of cytokines and other chemical mediators of inflammation (Fig. 33-10).

Acute pancreatitis is an inflammatory disorder believed to begin in the pancreas and is generally restricted to it; although in some cases its effects can be systemic, diverse and result in multiple organ failure. In 1896, Chiari advanced the understanding of acute pancreatitis by proposing the concept that the pancreatitis is essentially the premature, intrapancreatic activation of digestive enzymes, resulting in auto-digestion of the organ. Since then the intra-acinar activation of zymogens has been demonstrated consistently in multiple animal models of acute pancreatitis and is considered a central piece in the puzzle of acute pancreatitis.35,36 The key role of trypsin activation in acute pancreatitis has gained additional support from recent studies showing that mice lacking trypsinogen-7 (the isoform of trypsinogen which is activated during acute pancreatitis in mice) have significantly less pancreatic injury during acute pancreatitis37 and that intra-acinar expression of active trypsin causes pancreatitis in mice.38 The role of trypsin activation in the pathophysiology of acute pancreatitis has also been suggested by clinical studies; e.g., hereditary pancreatitis is associated with mutations that lead to elevated intracellular trypsin activation39 and activation of trypsinogen has been demonstrated in clinical pancreatitis as well.40
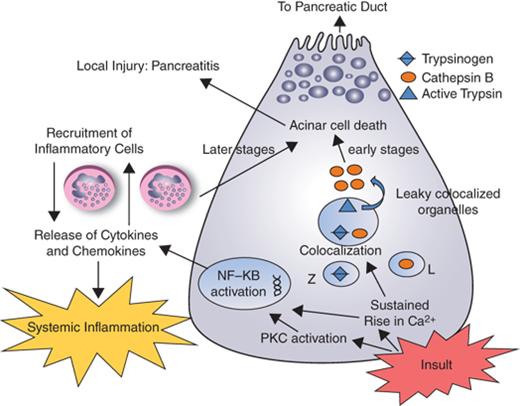
The mechanisms by which injurious stimuli lead to intra-acinar activation of trypsinogen and autodigestion of the gland have been the focus of research in pancreatitis for decades. Because the exocrine pancreas produces enzymes that are potentially injurious to it, several protective mechanisms have evolved to prevent autodigestion under normal conditions. Enzymes are synthesized as inactive precursors called proenzymes or zymogens, which are then transported and secreted outside the gland. Their activation occurs safely in the duodenum, where the brush-border enzyme enteropeptidase (or enterokinase) activates the trypsinogen, and the resulting trypsin then activates the other zymogens in a cascade reaction. This separates the site of production of these enzymes from the site of activation and thus the pancreas is insulated against enzymatic attack. Within the acinar cell itself, the potentially harmful digestive enzymes are segregated from the surrounding cytoplasm by being enclosed within membrane-bound organelles referred to as zymogen granules. Another layer of protection is provided by the synthesis of trypsin inhibitors, which are transported and stored along with the digestive enzyme zymogens. These are available to inhibit small amounts of prematurely-activated trypsinogen within the pancreatic acinar cells. It is theorized that acute pancreatitis occurs when this process goes awry and the gland is injured by the erroneously-activated enzymes that it produces. Although the mechanism(s) of erroneous activation are not fully understood, it has been shown that intra-acinar activation of trypsinogen goes hand-in-hand with inhibition of acinar secretion.41,42 Furthermore, in the presence of injurious stimuli, the zymogens responsible for initiating the disease are not secreted outside, but are observed to co-localize with cytoplasmic vacuoles that contain lysosomal enzymes such as cathepsin B.43 Data suggest that cathepsin B in these vacuoles activates trypsinogen. Thus, inhibition of cathepsin B by pharmacological inhibitors44 or by genetic deletion of cathepsin B eliminates trypsin activation and decreases the severity of pancreatitis in animal models.45 What leads to the coming together of zymogens and lysosomal hydrolases is unclear, but injurious stimuli leading to sustained cytosolic calcium increase have been indicted. Blocking this calcium increase prevents co-localization and activation of trypsin, and decreases injury due to pancreatitis.46 Based on these data, pre-ERCP supplementation of magnesium, a natural antagonist of calcium, is currently being evaluated as a strategy to decrease post-ERCP pancreatitis.47 How activation of trypsin in the co-localization vacuoles leads to pancreatic damage is also not clear. Recent work has led to the novel hypothesis that the lysosomal hydrolase cathepsin B activates trypsinogen to trypsin within the co-localization vacuoles. Trypsin then permeabilizes these co-localization vacuoles causing the release of cathepsin B into the cytosol. Once in the cytosol, cathepsin B initiates apoptotic cell death by permeabilizing mitochondrial membranes, which allows cytochrome C to be released into the cytosol. This initiates the apoptotic cascade and results in the apoptotic death of the acinar cells (Fig. 33-11).48
Figure 33-11.
Schematic representation of the acinar cell events in acute pancreatitis. When acinar cells are pathologically stimulated, their lysosomal (L) and zymogen (Z) contents colocalize, consequently trypsinogen is activated to trypsin by cathepsin B. Increased cytosolic calcium is required for colocalization. Once trypsin has permeabilized the contents of the cytosol, cathepsin B and other contents of these colocalized organelles are released. Once in the cytosol, cathepsin B activates apoptosis by causing cytochrome c to be released from the mitochondria. Activation of PKC results in a sudden activation of nuclear factor kappa beta (NKκb) which in turn triggers the release of cytokines that attract inflammatory response cells which mediate local and systemic inflammation cascades.37
Activated neutrophils are attracted to a focus of tissue injury and after activation release superoxides (the “respiratory burst”) and proteolytic enzymes (cathepsins, elastase and collagenase) which cause further injury. In addition macrophages release cytokines (including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-8) which mediate the local and the systemic inflammatory responses. These inflammatory mediators cause an increased pancreatic vascular permeability, leading to edema, hemorrhage, and microthrombi. Fluid may collect in and around the pancreas. The failure of the pancreatic microcirculation, a feature of more severe acute pancreatitis, results in pancreatic hypoperfusion and necrosis. Acute inflammation of the pancreatic parenchyma and peripancreatic tissues, but without recognizable necrosis is termed interstitital edematous pancreatitis.24 When necrosis is present, and evidenced by pancreatic hypoperfusion with constrast CT, it is termed necrotizing pancreatitis (Fig. 33-12). The updated morphological definitions and the contrast enhanced CT criteria for the diagnosis of the local complications of acute pancreatitis have recently been published in the revised Atlanta statement,49 and summarized in Table 33-5.50
| CONTENT | ACUTE (<4 WEEKS, NO DEFINED WALL) | CHRONIC (>4 WEEKS, DEFINED WALL) | ||
|---|---|---|---|---|
| No infection | Infection | No infection | Infection | |
| Fluid | Acute pancreatic fluid collection (APFC) | Infected APFC | Pseudocyst | Infected pseudocyst |
| Solid é fluid | Acute necrotic collection (ANC) | Infected ANC | Walled off necrosis (WON) | Infected WON |

An important aspect of the pathophysiology of acute pancreatitis is the mechanism by which events occurring in the pancreas induce systemic inflammation and multiorgan failure.The NFκB- dependent inflammatory pathway is one such key pathway. NFκB activation parallels trypsin activation in acute pancreatitis but appears to be independent of it. The role of trypsin in NFκB activation was debated for a long time, but this issue seems to be largely settled following the observation that NFκB activation still occurs in acini from trypsin knockout mice, which obviously do not have pathologic trypsin activation.37 However, sustained calcium increase, which leads to trypsinogen activation, is critical for NFκB activation as well, since attenuation of cytosolic calcium also abrogates NFκB activation.51 Once activated, NFκB regulates synthesis of multiple cytokines and chemokines, leading to recruitment of various inflammatory cells that then magnify and propagate systemic inflammation.
Although intra-acinar events initiate acute pancreatitis, events occurring subsequent to acinar cell injury determine the severity of pancreatitis. Once recruited to the pancreas, various inflammatory cells lead to further acinar cell injury and cause an elevation of various pro-inflammatory mediators such as TNF-α; IL-1, IL-2, IL-6, and other chemokines and anti-inflammatory factors.36 Elucidation of these mediators has encouraged efforts to target their production or activity with an aim to modulate the course of severe acute pancreatitis.
Organ failure can develop at any stage of acute pancreatitis, associated with an overwhelming proinflammatory response early, or later secondary to the development of infected local complications. The drivers of the systemic response are poorly understood, although factors include the elaboration of proinflammatory cytokines, and it appears that mesenteric lymph, bypassing the liver and containing these constituents, may contribute to the development of organ failure.52 The development of pancreatic necrosis, the breakdown of the intestinal barrier and the suppression of the immune response through the compensatory inflammatory response contribute to the development of infected pancreatic necrosis, the incidence of which peaks in the third to fourth week. This is usually associated with deterioration in the patient and may be associated with the late development of the systemic inflammatory response syndrome (SIRS) and multiorgan dysfunction syndrome/failure (MODS/F).
Organ failure is scored using the Marshall or Sequential Organ Failure Assessment (SOFA) systems (Table 33-6). The three organ systems most frequently involved are cardiovascular, respiratory, and renal. Multiple organ failure is defined as two or more organs registering 2 or more points on these scoring systems.49 Monitoring organ failure over time and in response to treatment is important in the clinical care of the disease.
| 0 | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| Respiration | |||||
| (PaO2FIO2) (mm Hg) | >400 | ≤400 | ≤300 | ≤200 with respiratory support | ≤100 with respiratory support |
| Coagulation | |||||
| Platelets (xl01 per μL) | >150 | ≤150 | ≤100 | ≤50 | ≤20 |
| Liver | |||||
| Bilirubin (μmol/L) | <20 | 20–32 | 33–101 | 102–204 | >204 |
| Cardiovascular | |||||
| Hypotension | No hypotension | MAP <70mm Hg | Dopamine ≤5 or dobutamine (any dose)∗ | Dopamine >5 or epi ≤0.1∗ or norepi ≤0.1∗ | Dopamine >15 or epi >0.1∗ or norepi >0.1∗ |
| Central nervous system | |||||
| Glasgow coma score | 15 | 13–14 | 10–12 | 6–9 | <6 |
| Kidney | |||||
| Creatinine (μmol/L) or urine output | <110 | 110–170 | 171–299 | 300–440 or <500 ml/day | >440 or <200 ml/day |
The management of acute pancreatitis covers a wide spectrum of severity. Patients with suspected acute pancreatitis should be admitted to hospital. Those with mild acute pancreatitis usually remain in hospital for less than a week, while those with severe and critical acute pancreatitis may require many weeks or months of intensive treatment. The risk of mortality reflects this spectrum of severity. The risk is less than 1% for those with mild disease, increasing to around 10% for those with moderate disease, but for severe and critical disease the mortality risk is much higher (20% to 40% and greater than 50%, respectively). The earlier identification of these high-risk categories and the transfer of these patients to specialized centers is an important priority of management.53 Scoring systems such as Ranson’s Criteria identify these high-risk patients (Table 33-7). The management of acute pancreatitis should be multidisciplinary and it is important that a coordinated care plan is carefully supervised.
| Criteria for acute pancreatitis not due to gallstones | |
| At admission | During the initial 48 h |
| Age >55 y | Hematocrit fall >10 points |
| WBC >16,000/mm3 | BUN elevation >5 mg/dL |
| Blood glucose >200 mg/dL | Serum calcium <8 mg/dL |
| Serum LDH >350 IU/L | Arterial PO2 <60 mm Hg |
| Serum AST >250 U/dL | Base deficit >4 mEq/L |
| Estimated fluid sequestration >6 L | |
| Criteria for acute gallstone pancreatitis | |
| At admission | During the initial 48 h |
| Age >70 y | Hematocrit fall >10 points |
| WBC >18,000/mm3 | BUN elevation >2 mg/dL |
| Blood glucose >220 mg/dL | Serum calcium <8 mg/dL |
| Serum LDH >400 IU/L | Base deficit >5 mEq/L |
| Serum AST >250 U/dL | Estimated fluid sequestration >4 L |
The diagnosis of acute pancreatitis requires the patient to present with abdominal pain consistent with acute pancreatitis (acute onset of a severe constant epigastric pain which often radiates through to the mid back) and the elevation of serum amylase or lipase (>3 times upper limit of normal). Imaging (usually by contrast enhanced CT scanning) is only required for the diagnosis of acute pancreatitis when these diagnostic criteria are not met.49 Because of the many causes of hyperamylasemia, it is important to measure either the pancreatic isoenzyme of amylase or lipase.
The serum amylase concentration increases almost immediately with the onset of disease and peaks within several hours.54 It remains elevated for 3 to 5 days before returning to normal. There is no significant correlation between the magnitude of serum amylase elevation and severity of pancreatitis; in fact, a milder form of acute pancreatitis is often associated with higher levels of serum amylase compared with that in a more severe form of the disease.
It is important to note that hyperamylasemia can also occur as a result of conditions not involving pancreatitis. For example, hyperamylasemia can occur in a patient with small bowel obstruction, perforated duodenal ulcer, or other intra-abdominal inflammatory conditions. In contrast, a patient with acute pancreatitis may have a normal serum amylase level, which could be due to several reasons. In patients with hyperlipidemia, values might appear to be normal because of interference by lipids with chemical determination of serum amylase. In many cases, urinary clearance of pancreatic enzymes from the circulation increases during pancreatitis; therefore, urinary levels may be more sensitive than serum levels. For these reasons, it is recommended that amylase concentrations also be measured in the urine. Urinary amylase levels usually remain elevated for several days after serum levels have returned to normal. In patients with severe pancreatitis associated with significant necrotic damage, the pancreas may not release large amounts of enzymes into the circulation.
With increasing severity of disease, the intravascular fluid loss may become life-threatening as a result of sequestration of edematous fluid in the retroperitoneum. Hemoconcentration then results in an elevated hematocrit. However, there also may be bleeding into the retroperitoneum or the peritoneal cavity. In some patients (about 1%), the blood from necrotizing pancreatitis may dissect through the soft tissues and manifest itself as a bluish discoloration around the umbilicus (Cullen’s sign) or in the flanks (Grey Turner’s sign). The severe fluid loss may lead to prerenal azotemia with elevated blood urea nitrogen and creatinine levels. There also may be hyperglycemia, hypoalbuminemia, and hypocalcemia sufficient in some cases to produce tetany.
Pain is the cardinal symptom of acute pancreatitis and its relief is a clinical priority. There is a lack of high quality evidence to guide the choice of analgesic. Because of unpredictable absorption analgesia should be administered intravenously, at least at the outset and before oral intake has been established. Those with mild pain can usually be managed with a nonsteroidal anti-inflammatory drug (e.g., metamizole 2g/8h IV) while those with more severe pain are best managed with opioid analgesia (e.g., buprenorphine 0.3mg/4h IV). Administration of buprenorphine, pentazocine, procaine hydrochloride, and meperidine are all of value in controlling abdominal pain. Morphine is to be avoided, due to its potential to cause sphincter of Oddi spasm.
Where classification relates to the present or past severity of acute pancreatitis, prediction is about the future and ultimate severity and outcome of the patient. Accurately predicting acute pancreatitis severity is important in making triage decisions about whether a patient should be transferred to a tertiary hospital, or admitted to an intensive care unit, and in making decisions about fluid therapy, whether an ERCP is indicated, and other issues. There is a long history of attempts to find prognostic or predictive markers that accurately stratify the risk, with the most widely used being the Ranson’s criteria (Table 33-7) or modified Glasgow criteria. Both use clinical and biochemical parameters scored over the first 48 hours of admission. When there are 3 or more positive criteria, the disease is considered “predicted severe.” There are many other approaches to predicting severity. At 24 hours after admission an APACHE II score of 8 or more or a serum C-reactive protein level of >150mg/dl has a similar accuracy in predicting severity as Ranson’s criteria.54 The more recently proposed Bedside Index for Severity of Acute Pancreatitis (BISAP) is calculated from blood urea nitrogen (> 25 mg/dl), impaired mental status (GCS <15), presence of systemic inflammatory response syndrome (SIRS), age >60 years, and pleural effusion. Although it has the advantage of simplicity and can be performed within the first 24 hours of admission it has performed no better than other predictors in comparison studies.55 The presence of SIRS also has prognostic significance.56 There remains some controversy as to the importance of obesity as a risk factor for severe and critical acute pancreatitis.57 Another approach has been taken in seeking to predict those with ‘harmless’ acute pancreatitis58 using three factors that can be determined on admission; absence of rebound tenderness or guarding, normal hematocrit, and normal serum creatinine. The accuracy of this approach appears to be over 90%, and triages most patients with acute pancreatitis away from intensive care.
Unfortunately these and many other single and combined predictors of severity have an accuracy of only around 70%.54 This means that there is misclassification error of 30% that limits the value in predicting the severity of acute pancreatitis in individual patients. In the absence of any new biomarkers of ultimate disease severity, making better use of existing predictors through sequencing tests, combining tests or using artificial neural network methodologies has shown promise.59
Accurately classifying or staging acute pancreatitis severity is important for clinical decision-making, communication, and enrollment into trials. The wide spectrum of pancreatitis severity has not been captured in the previous binary classifications (mild or severe). A four-category classification of severity (mild, moderate, severe, critical) (Table 33-8) has been developed on the principle of casual inference, derived by meta-analysis, refined by an international multidisciplinary process and independently validated.60 The key determinants of severity are local complications (absent, sterile or infected) and systemic complications (absent, transient organ failure, or persistent organ failure).61The tracking of patient severity is important in delineating the clinical trajectory of a patient, and this classification of severity can be applied on a frequent basis. It can also be used in retrospect for audit purposes.
| DETERMINANTS | NO LOCAL COMPLICATIONS | STERILE LOCAL COMPLICATIONS | INFECTED LOCAL COMPLICATIONS |
|---|---|---|---|
| NO ORGAN FAILURE | MILD | MODERATE | SEVERE |
| TRANSIENT ORGAN FAILURE | MODERATE | MODERATE | SEVERE |
| PERSISTENT ORGAN FAILURE | SEVERE | SEVERE | CRITICAL |
A history of alcohol ingestion must be ascertained and preferably confirmed with blood ethanol levels. Gallstones should be investigated by ultrasonography. A gallstone etiology is more likely in females over the age of 50 with an elevation of alkaline phosphatase (>300 IU/L), alanine transferase (>100 IU/L) and amylase (>4000 IU/L). In the absence of gallstones and alcohol, a systematic approach to the identification of another factor will include taking a history for drug use, trauma, ERCP, infection, and measuring serum triglycerides, calcium, and others (Table 33-4).
1. Diagnosis • History of abdominal pain consistent with acute pancreatitis • >3x elevation of pancreatic enzymes • CT scan if required to confirm diagnosis 2. Initial assessment / management (first 4 hrs) • Analgesia • Fluid resuscitation • Predict severity of pancreatitis ° Ranson’s criteria ° HAPS score • Assess systemic response ° SIRS score ° SOFA (organ failure) 3. Reassessment / management (4 to 6 hrs) • Assess response to fluid resuscitation ° mean arterial pressure ° heart rate ° urine output ° hematocrit • Determine etiology ° Ultrasound for gallstones/sludge ° History of alcohol consumption ° Laboratory evaluation of other causes • MRCP and/or Urgent ERCP if concomitant cholangitis is present ° not for cholestasis or predicted severe disease per se • Transfer to ICU or specialist center as needed ° Deterioration or failure to respond to initial management ° Intensive support for persistent organ failure • Commence enteral nutrition ° Once normovolemia restored (usually after 6 hours) ° Commence via NG tube if no gastric stasis • No prophylactic antibiotics or probiotics | 4. Conservative management and monitoring (at least daily) • Clinical evaluation ° Assess cardiovascular, respiratory, and renal function ° Detect peritonitis and abdominal compartment syndrome • Daily C-Reactive Protein • Classify severity (mild, moderate, severe, critical) • Detect intolerance of NG EN ° Advance tube for NJ feeding if needed ° Consider supplemental Parenteral Nutrition by day 4 5. Indications for “pancreatic protocol CT scan” (rarely in 1st week) • For significant clinical deterioration and elevated CRP • For suspicion of local pancreatic complications • For suspected bowel ischemia • For acute bleeding (CTa) (if stable enough & consider embolization) • For abdominal compartment syndrome 6. Invasive intervention • For deteriorating patient with suspected infected local complication • “Step up approach” with initial drain guided by current CT scan (percutaneous or endoscopic drainage) • Delay for 3 to 4 weeks with intensive care support, if possible • If failure to respond or secondary deterioration, repeat CT scan, and select appropriate minimally invasive technique based on available expertise and equipment ° Video-assisted retroperitoneal debridement or percutaneous nephroscopic debridement ° Endoscopic transluminal debridement ° Ongoing large bore drainage and irrigation 7. Indication for laparotomy • Failed “step-up approach” for further debridement/drainage • Acute abdomen (perforation or ischemia) • Severe abdominal compartment syndrome (rarely) |
Fluid therapy to restore and maintain circulating blood volume is the most important intervention in the early management of acute pancreatitis.62 A recent systematic review has shown that the evidence base for fluid therapy is scant, however.63 It is not known which fluid to give, how aggressively to administer it or what goal to use as a guide or to monitor the response to it. While there are proponents for aggressive fluid therapy and for specific resuscitation goals, it is probably best to resuscitate with a balanced crystalloid and to restore normal blood volume, blood pressure, and urine output. On the basis of recent data it appears that lactated Ringer’s solution may be superior to normal saline in reducing the systemic inflammatory response.64 Caution needs to be exercised in those with cardiac and renal disease and in the elderly.
In contrast to analgesia and fluid therapy there is a sound evidence base for nutritional support in acute pancreatitis.65 It is no longer acceptable to “rest the pancreas” by avoiding enteral nutrition, and parenteral nutrition should only be offered if the patient’s calculated nutritional requirements cannot be achieved by the enteral route. Enteral nutrition should be commenced after initial fluid resuscitation and within the first 24 hours of admission. It can be introduced through a nasogastric tube and increased in step-wise fashion over 2 to 3 days.66 The tube can be advanced to the jejunum, by endoscopy or fluoroscopy, if there is evidence of feeding intolerance. A delay in commencing enteral nutrition may contribute to the development of intestinal ileus and feeding intolerance. Aggressive early enteral feeding, particularly prior to adequate resuscitation, may put the patient at risk of nonocclusive mesenteric ischemia. There is no evidence to support the use of elemental or immune-enhancing formulas over standard polymeric formulas.67
In predicted mild acute pancreatitis the transition from oral fluids to food is usually timed to when the patient’s abdominal pain has resolved, but the trend is toward allowing patients to resume intake ad libidum (i.e., patient controlled nutrition).
It may be necessary to perform a CT scan to diagnose acute pancreatitis in patients who are severely ill, or in those presenting with undifferentiated abdominal pain. But there is no advantage in using CT findings, or a “CT Severity Index,” over other methods in predicting the severity of acute pancreatitis.54 The primary purpose of cross sectional imaging is the diagnosis of local complications, in particular the development and extent of pancreatic necrosis or the presence of different fluid collections. CT scanning is important to guide the insertion of percutaneous drains in the management of local complications. MR is superior to CT scanning in detecting any solid content within collections (Fig. 33-13). When bleeding is suspected in association with a local complication, an arterial phase CT scan (CTa) is useful in detecting a pseudoaneurysm, active bleeding, and/or hematoma.
Figure 33-13.
Corresponding CT (A) and MR (B) images of a patient with a symptomatic pseudocyst. CT image reveals a well circumscribed homogenous collection (arrows) exerting mass effect on antrum of stomach. The T2-weighted MR image clearly distinguishes necrotic pancreas (black arrows) from fluid (white arrows). (Reproduced with permission from Bollen TL et al. Imaging of acute pancreatitis: Update of the revised Atlanta Classification. Radiol Clin North Am. 2012;50:429. Copyright Elsevier.)
Randomized trials have demonstrated that early ERCP (within 24 or 48 hours of admission) reduces complications, but not mortality, in patients with predicted severe gallstone-associated acute pancreatitis. The benefits of this invasive modality (release of the presumed impacted stone) may be offset, however, but the risks of the procedure, which include an increase in the severity of pancreatitis, bleeding, cholangitis, and duodenal perforation. More recent evidence has suggested that early ERCP confers no benefit in the absence of concomitant cholangitis, as the offending common duct stone usually passes before ERCP can be performed.68 If symptoms and chemical abnormalities persist or increase after intial presentation, magnetic resonance cholangio-pancreatography (MRCP) can detect a filling defect in the distal common bile duct, and may be used as prerequisite for attempted ERCP.69 Persistent cholestasis without cholangitis may require an ERCP but is rarely indicated in the acute setting.
Although the use of broad-spectrum antibiotics to treat established infection in acute pancreatitis is a well-established practice, there has been considerable controversy surrounding the use of prophylactic antibiotics. The overuse of antibiotics has been associated with a documented rise in fungal infections and resistant organisms. Overall, it appears that the most recent and generally better designed studies do not support the use of prophylactic antibiotics to reduce the frequency of pancreatic infectious complications, surgical intervention, and death.70
Vigilance is required for the timely and accurate diagnosis of local complications. The decisions regarding how and when to intervene are often difficult. While guided by the information gained by cross-sectional imaging, the decision to intervene is based on the clinical status and trajectory of the patient and the poor response to maximal intensive care support. This means close monitoring of the patient by serial examination, supplemented by regular measurement of inflammatory markers (e.g., C reactive protein) and a pancreatic protocol CT scan if a local complication is suspected and intervention considered warranted. In practice, intervention is delayed in order to allow demarcation of necrosis, and a reduced the risk of bleeding, disseminated infection and collateral damage to adjacent organs by an intervention (Fig. 33-14). Appreciation of this has resulted in a notable trend toward delayed intervention, now uncommon before 3 to 4 weeks from the onset of symptoms. An important emerging approach is the increasing use of percutaneous catheter drainage in patients with suspected infected complications.71 Fine needle aspiration is now rarely used to confirm infection because the insertion of a needle at the time of planned drainage allows confirmation of the suspected infection. Preemptive drainage with one or more catheters often produces improvement or stabilization of the patient’s overall clinical status.72 In this way drainage “buys time” and allows the lesion to become more walled off and safer to treat. Recent data suggest that primary percutaneous catheter drainage may be the only intervention required in a third to a half of patients and that this proportion might increase further if there were a standard protocol of regular catheter exchange, upsizing and irrigation.72 A proportion of patients do however require further treatment when they fail to respond (the so-called “step up approach”) and there is a wide array of minimally invasive options to choose from73(Fig. 33-15). These interventions can be classified on the basis of the method of visualization, route taken to the lesion and the purpose of the intervention.74 In practice the approach taken will depend on local expertise and equipment as well as the location and type of the specific local complication. A large Dutch randomized trial has shown that open surgical techniques should only be considered in those who fail to respond to the step-up approach, that is prior percutaneous drainage and minimally invasive intervention.75 The exception is to be found in the rare situation where an abdominal compartment syndrome requires open decompression, but this is usually earlier than the optimal time to intervene for local complications. A recent landmark randomized trial has compared two minimally invasive techniques, endoscopic transgastric drainage and the videoscope assisted retroperitoneal debridement through a flank incision (Fig. 33-15). The data show that the former approach is superior, although the latter has a role when the walled off necrosis is remote from the stomach or duodenum, as in the left flank.76
The management of an acute noninfected pseudocyst is usually conservative, as about half of these will resolve spontaneously. When symptoms of pain or inability to eat persist or infection occurs, intervention is required. Pseudocysts persist because of communication with the main pancreatic duct and/or distal ductal stenosis. Percutaneous drainage should be avoided in this situation because of the risk of external pancreatic fistula.77 EUS guided internal drainage into stomach, duodenum, or transpapillary pancreatic duct stenting is the preferred approach.
Figure 33-15.
Two minimally invasive interventions for local complications of acute pancreatitis: A. video-assisted retroperitoneal debridement and B. endoscopic transgastric necrosectomy 76. (Reproduced with permission from Bakker OJ et al. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: A randomized trial. JAMA. 2012;307(10):1053. Copyright © 2012 American Medical Association. All rights reserved.)
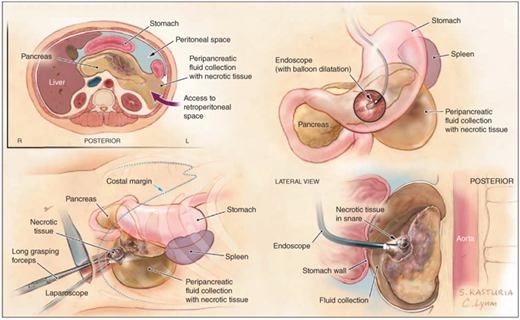
The specific management of multiple organ failure is beyond the scope of this chapter. The early identification of organ dysfunction and failure is important because it is a key determinant of severity and outcome and to facilitate the timely transfer of the patient to an intensive care unit to optimize management, provide organ support, and allow more intensive monitoring. The severity of organ failure can be scored (Table 33-6). The responsiveness of organ failure to resuscitation over the first 48 hours is an important prognostic clue; those that respond have transient organ failure and have a better outlook than those who do not respond and have persistent organ failure.78 Organ failure that develops later in the disease course is usually secondary to infection of a local complication and should be managed accordingly (Table 33-9).
While it is widely accepted that cholecystectomy is essential to prevent gallstone pancreatitis recurrence, the timing of cholecystectomy is important. Index cholecystectomy, done in the same admission and prior to discharge, appears safe and can almost always be accomplished laparoscopically.79 But index cholecystectomy is not suitable for all patients, particularly some who have had local pancreatic complications which includes a large phlegmon which extends into the porta hepatis. These patients may require an interval cholecystectomy after resolution of the inflammatory process. If surgery is required for the management of local complications, then a cholecystectomy is often performed at that time.
The management of acute pancreatitis remains a formidable challenge due to the variety and severity of many associated complications (Table 33-10), and continues to evolve. Although specific treatments for acute pancreatitis remain elusive, progress has been made in the management of pain, fluid resuscitation, antibiotic prophylaxis, enteral nutrition, therapeutic ERCP, and cholecystectomy. Progress has also been made in the intensive care management of systemic complications and in the development of less invasive interventions for the treatment of local complications, particularly infected pancreatic necrosis.
I. Local A. Pancreatic phlegmon B. Pancreatic abscess C. Pancreatic pseudocyst D. Pancreatic ascites E. Involvement of adjacent organs, with hemorrhage, thrombosis, bowel infarction, obstructive jaundice, fistula formation, or mechanical obstruction II. Systemic A. Pulmonary 1. Pneumonia, atelectasis 2. Acute respiratory distress syndrome 3. Pleural effusion B. Cardiovascular 1. Hypotension 2. Hypovolemia 3. Sudden death 4. Nonspecific ST-T wave changes 5. Pericardial effusion C. Hematologic 1. Hemoconcentration 2. Disseminated intravascular coagulopathy D. GI hemorrhage 1. Peptic ulcer 2. Erosive gastritis 3. Portal vein or splenic vein thrombosis with varices E. Renal 1. Oliguria 2. Azotemia 3. Renal artery/vein thrombosis F. Metabolic 1. Hyperglycemia 2. Hypocalcemia 3. Hypertriglyceridemia 4. Encephalopathy 5. Sudden blindness (Purtscher’s retinopathy) G. Central nervous system 1. Psychosis 2. Fat emboli 3. Alcohol withdrawal syndrome H. Fat necrosis 1. Intra-abdominal saponification 2. Subcutaneous tissue necrosis |
CHRONIC PANCREATITIS
Chronic pancreatitis is an incurable, chronic inflammatory condition that is multifactorial in its etiology, highly variable in its presentation, and a challenge to treat successfully. Autopsy studies indicate that evidence of chronic inflammation, such as fibrosis, duct ectasia, and acinar atrophy is seen in up to 5% of the population,80 although these data are difficult to interpret because many of these changes are also present in asymptomatic elderly patients.81 Population studies suggest a prevalence that ranges from 5 to 40 persons per 100,000 population, with considerable geographic variation.82 Differences in diagnostic criteria, regional nutrition, alcohol consumption, and medical access account for variations in the frequency of the diagnosis, but the overall incidence of the disease has risen progressively over the past 50 years.
There are multiple etiologies of chronic pancreatitis, including genetic mutations, alcohol exposure, duct obstruction due to trauma, gallstones, and tumors, metabolic diseases such as hyperlipidemia and hyperparathyroidism, and auto-immune disease. In addition, nutritional causes include so-called “tropical pancreatitis,” which has been thought to result from ingestion of certain starches. A significant number of patients have no discernible cause of the disease despite extensive testing, and are said to have idiopathic chronic pancreatitis.
In 1952, Comfort and Steinberg reported a kindred of “hereditary chronic relapsing pancreatitis” after treating the proband, a 24-year-old woman, at the Mayo Clinic.83 Subsequently, familial patterns of chronic, nonalcoholic pancreatitis have been described worldwide, and a familiar pattern has emerged. Typically, patients first present in childhood or adolescence with abdominal pain and are found to have chronic calcific pancreatitis on imaging studies. Progressive pancreatic dysfunction is common, and many patients present with symptoms due to pancreatic duct obstruction. The risk of subsequent carcinoma formation is increased, reaching a prevalence, in some series, of 40%, but the age of onset for carcinoma is typically >50 years old.84 The disorder is characterized by an autosomal dominant pattern of inheritance, with 80% penetrance and variable expression. The incidence is equal in both sexes.
Whitcomb and colleagues,85 and separately LeBodic and associates,86 performed gene linkage analysis and identified a linkage for hereditary pancreatitis to chromosome 7q35. Subsequently, the region was sequenced and revealed eight trypsinogen genes. Mutational analysis revealed a missense mutation resulting in an Arg to His substitution at position 117 of the cationic trypsinogen gene, or PRSS1, one of the primary sites for proteolysis of trypsin. This gain-of-function mutation results in an excess production of trypsinogen, which results in persistent and uncontrolled proteolytic activity and autodestruction within the pancreas.87 The position 117 mutation of PRSS1 and an additional mutation, now known collectively as the R122H and N291 mutations of PRSS1, account for about two-thirds of cases of hereditary pancreatitis. Recently, Masson and associates described a gain-of-function mutation in the anionic trypsinogen gene, PRSS2, that is also present in some cases.88
Similarly,SPINK1, an inflammation-induced trypsin inhibitor secreted in acinar cells, has been found to have a role in hereditary pancreatitis. SPINK1 specifically inhibits trypsin action by competitively blocking the active site of the enzyme. Witt and colleagues investigated 96 unrelated children with chronic pancreatitis in Germany and found a variety of SPINK1 mutations in 23% of the patients.89 Several studies have now confirmed an association of loss-of-function SPINK1 mutations with familial and idiopathic forms of chronic pancreatitis, as well as so-called tropical pancreatitis.90,91 SPINK1 mutations are common in the general population as well, and the frequency of these mutations varies in different cohorts of idiopathic chronic pancreatitis, from 6.4% in France92 to 25.8% in the United States.93 Thus, hereditary pancreatitis results from one or more mutational defects that incapacitate an auto-protective process that normally prevents proteolysis within the pancreas.
Cystic fibrosis, originally termed cystic fibrosis of the pancreas, results from a variety of mutations of the cystic fibrosis transmembrane receptor (CFTR). The CFTR is present in pancreatic duct cells, and controls the amount of chloride and bicarbonate secreted into the normally alkaline pancreatic juice. The CFTR gene contains over 4300 nucleotides, divided into 24 exons, which encode a 1480-amino acid protein. Over 1000 polymorphisms have been reported, and many are common.32 The CFTR mutation associated with the classic pulmonary disease, F508, is rarely observed in chronic pancreatitis. But other CFTR mutations have been noted to be associated with chronic idiopathic pancreatitis and auto-immune pancreatitis in which the pulmonary, intestinal, and cutaneous manifestations of the disease are silent.94
Many studies have been undertaken to determine whether specific genetic abnormalities are associated with alcoholic chronic pancreatitis, and which might confer susceptibility to the disease. A 5.8% rate of SPINK1 mutations in patients with alcoholic pancreatitis has been shown, compared to a 0.8% rate in the control population, and studies of the known mutations of the CFTR gene have failed to demonstrate a strong association with alcoholic chronic pancreatitis.89
In 2012, a landmark study by Whitcomb and associates demonstrated a likely genetic cause of the predisposition to alcohol-induced chronic pancreatitis in men95. In a genome-wide association study of more than 2000 patients, these researchers discovered that a common DNA variant on the X chromosome is present in 26% of men without pancreatitis, but jumps to nearly 50% of men diagnosed with alcoholic pancreatitis. The variant involves the claudin 2 (CLDN2) gene, which encodes a tight junction protein normally present in ductal cells. In cases of chronic pancreatitis, the CLDN2 protein is abnormally expressed in acinar cells, and may alter the secretory dynamics of enzyme release. The abnormality does not appear to cause pancreatitis but if pancreatitis occurs for any reason in a person with the CLDN2 variant, it is more likely that the person will develop chronic pancreatitis; the risk is increased even further among alcohol users. Only 10% of women have the X chromosome-linked variant on both X chromosomes, and most women with the CLDN2 variant on one X chromosome appear to be protected from alcoholic chronic pancreatitis by the other X chromosome, if it is normal. Men, with only one X chromosome, have no protection if they inherit a CLDN2 mutation. This helps to explain the high prevalence of alcoholic chronic pancreatitis among men, although the mechanism remains unclear. This study does not demonstrate a genetic cause for all cases of alcohol-related chronic pancreatitis, but shows that a genetic element contributes to many patients with the disease (Fig. 33-16).
Figure 33-16.
Schematic model of genetic causes of chronic pancreatitis. A pancreatic acinus is portrayed showing the pathway for digestive enzymes (zymogens) to be secreted by the acinar cell into the ductal system where water, sodium, and bicarbonate are secreted by the duct and centroacinar (CA) cells. Mutations in at least five genes have been identified as risk factors for chronic pancreatitis: gain-of function mutations in the cationic trypsinogen genes (PRSS1, PRSS2) cause hereditary pancreatitis. Cationic and anionic trypsinogen are normally active in the duodenum by enterokinase (EK). Premature activation of the trypsin in the acinar cell leads to zymogen activation, local cellular injury, and inflammation. Mutations of the trypsin inhibitors SPINK1 or chymotrypsin C (CTRC), or of the calcium sensing receptor CASR, result in premature activation of trypsinogen. Mutations in the cystic fibrosis transmembrane receptor CFTR results in trypsinogen stasis within the ducts due to insufficient secretion by duct cells. A Mutation in claudin-2 (CLDN2) results in abnormal expression in acinar cells, instead of its normal location between duct cells, and is associated with the accelerated development of chronic pancreatitis in alcohol abusers. (Adapted with permission from Macmillan Publishers, Ltd. Whitcomb DC et al: Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44:1349. Copyright © 2012.)
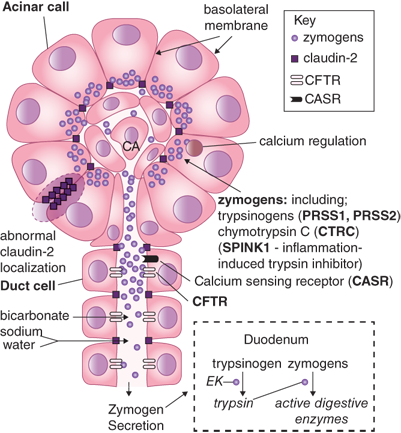
In 1878, Friedreich proposed that “a general chronic interstitial pancreatitis may result from excessive alcoholism (drunkard’s pancreas).”96 Since that observation, numerous studies have shown that a causal relationship exists between alcohol and chronic pancreatitis, but the prevalence of alcohol as the etiology of the disease in Western countries ranges widely, from 38% to 94%97(Fig. 33-17).
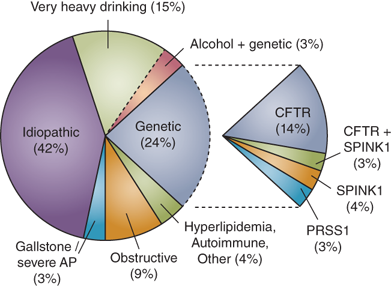
There is a linear relationship between exposure to alcohol and the development of chronic pancreatitis.98 The risk of disease is present in patients with even a low or occasional exposure to alcohol (1 to 20 g/d), perhaps due to the CLDN2 gene mutation described above, so there is no threshold level of alcohol exposure below which there is no risk of developing chronic pancreatitis. Furthermore, although the risk of disease is dose related, and highest in heavy (>150 g/d) drinkers, the prevalence of chronic pancreatitis among confirmed alcohol abusers is only 5% to15%.99 However, the duration of alcohol consumption is definitely associated with the development of pancreatic disease. The onset of disease typically occurs between ages 35 to 40 years, after 16 to 20 years of heavy alcohol consumption. Recurrent episodes of acute pancreatitis are typically followed by chronic symptoms after 4 or 5 years.100
Although the pattern of disease presentation is well known in those alcohol users who develop pancreatic disease, the pathophysiology of alcohol-induced pancreatic disease is still an area of active investigation. In their 1946 classic study, Comfort, Gambrill, and Baggenstoss proposed that chronic pancreatitis was the result of multiple episodes of acute inflammation, with residual and progressively increasing chronic inflammation.101 Subsequently, other investigators proposed that initial acute inflammation was not necessarily linked to chronic changes in the pancreas,102 and Kondo and associates showed that other, additional factors were necessary for repeated exposure to alcohol to cause chronic pancreatitis.103 Regardless of the requirement for other predisposing or facilitative factors, the concept that multiple episodes (or a prolonged course) of pancreatic injury ultimately leads to chronic disease is widely accepted as the pathophysiologic sequence104 (Fig. 33-18).
Figure 33-18.
“Multiple hit” theory of the etiology of chronic pancreatitis. Multiple episodes of acute pancreatitis cause progressively more organized inflammatory changes that ultimately result in chronic inflammation and scarring. [Reproduced with permission from Apte et al.104 Copyright Elsevier.]
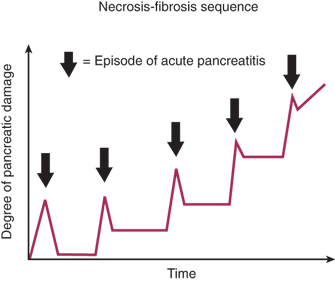
Although direct alcohol exposure to the pancreatic ductal system, or elevated levels of alcohol in the bloodstream, has been shown to alter the integrity and function of pancreatic ducts and acini directly,105 most investigators believe that alcohol metabolites such as acetaldehyde, combined with oxidant injury, result in local parenchymal injury that is preferentially targeted to the pancreas in predisposed individuals. Repeated or severe episodes of toxin-induced injury activate a cascade of cytokines, which, in turn, induces pancreatic stellate cells (PSCs) to produce collagen and cause fibrosis (Fig. 33-19). It remains to be determined whether alcohol sensitizes the pancreas of susceptible individuals to another cause of acute inflammation, or whether genetic or other factors predispose to direct alcohol-related injury.104
Figure 33-19.
The sentinel acute pancreatitis event (SAPE) hypothesis for the development of chronic pancreatitis. A critical episode of acute pancreatitis activates cytokine-induced transformation of pancreatic stellate cells, which results in collagen production and fibrosis. ETOH = ethyl alcohol. (Reproduced with permission from Schneider et al.135 Copyright Elsevier.)
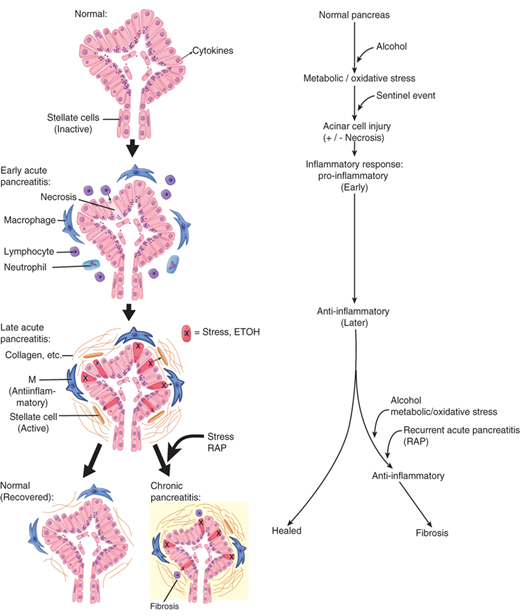
Alcohol may interfere with the intracellular transport and discharge of digestive enzymes, and may contribute to the colocalization of digestive enzymes and lysosomal hydrolase within acinar cells, leading to autodigestion106,107 (see section on Acute Pancreatitis). A high-protein, low-bicarbonate, low-volume secretory output is seen after chronic alcohol exposure which may contribute to the precipitation of proteins in secondary ducts in the early stages of chronic pancreatitis.108 Calcium is complexed to protein plugs in small ductules, secondary ducts, and, eventually, in the main ductal system, which causes ductal cell injury and obstruction of the secretory system, which further promotes an inflammatory response.
Cigarette smoking has been strongly associated with chronic pancreatitis,109 but until recently it was unclear whether this was a causative risk factor. Studies have now shown that smoking actually accelerates the development of alcoholic pancreatitis,110 and the risk of cancer in chronic pancreatitis is increased significantly by smoking. In hereditary pancreatitis, smoking has been found to lower the age of onset of carcinoma by about 20 years.111 Smoking therefore appears to be an independent risk factor for the late complications of alcoholic pancreatitis, if not an early cofactor for the development of fibrosis.
Hypercalcemia is a known cause of pancreatic hypersecretion,112 and chronic hypercalcemia caused by untreated hyperparathyroidism is associated with chronic calcific pancreatitis.113 Hypercalcemia is also a stimulant for pancreatic calcium secretion, which contributes to calculus formation and obstructive pancreatopathy. The treatment is correction of the hyperparathyroidism and assessment of any additional endocrinopathies.
In addition to the risk of acute pancreatitis, hyperlipidemia and hypertriglyceridemia predispose women to chronic pancreatitis when they receive estrogen replacement therapy.114 Fasting triglyceride levels <300 mg/dL are below the threshold for this to occur, and the mechanism of estrogen potentiation of hyperlipidemia-induced chronic pancreatitis is unknown. It is assumed that chronic changes occur after repeated subclinical episodes of acute inflammation. Aggressive therapy of hyperlipidemia is therefore important in peri- or postmenopausal patients who are candidates for estrogen therapy.
A major impediment to a better understanding of the etiology, frequency and severity of chronic pancreatitis has been the difficulty with which investigators and clinicians have struggled to identify a useful classification system. Multiple classification systems have been proposed. The TIGAR-O scheme categorizes chronic pancreatitis according to risk factors and etiologies, such as a) toxic-metabolic, b) idiopathic, c) genetic, d) auto-immune, e) recurrent and severe acute pancreatitis, or f) obstructive.115 A recent classification system based on histopathology as well as etiology, as delineated by Singer and Chari,116 is shown in Table 33-11.
| CHRONIC CALCIFIC PANCREATITIS | CHRONIC OBSTRUCTIVE PANCREATITIS | CHRONIC INFLAMMATORY PANCREATITIS | CHRONIC AUTOIMMUNE PANCREATITIS | ASYMPTOMATIC PANCREATIC FIBROSIS |
|---|---|---|---|---|
| Alcohol | Pancreatic tumors | Unknown | Associated with autoimmune disorders (e.g., primary sclerosing cholangitis) | Chronic alcoholic |
| Hereditary | Ductal stricture | Endemic in asymptomatic residents in tropical climates | ||
| Tropical | Gallstone- or trauma-induced or pancreas divisum | |||
| Hyperlipidemia | Sjögren’s syndrome | |||
| Hypercalcemia | Primary biliary cirrhosis | |||
| Drug-induced | ||||
| Idiopathic |
This type is the largest subgroup in the classification scheme proposed by Singer and Chari, and includes patients with calcific pancreatitis of most etiologies. Although the majority of patients with calcific pancreatitis have a history of alcohol abuse, stone formation and parenchymal calcification can develop in a variety of etiologic subgroups; hereditary pancreatitis and tropical pancreatitis are particularly noteworthy for the formation of stone disease. The clinician should therefore avoid the assumption that calcific pancreatitis confirms the diagnosis of alcohol abuse.
This refers to chronic inflammatory changes that are caused by the compression or occlusion of the proximal ductal system by tumor, gallstone, posttraumatic scar, or inadequate duct caliber (as in pancreas divisum). Obstruction of the main pancreatic duct by inflammatory (posttraumatic) or neoplastic processes can result in diffuse fibrosis, dilated main and secondary pancreatic ducts, and acinar atrophy. The patient may have little in the way of pain symptoms or may present with signs of exocrine insufficiency. Intraductal stone formation is rare, and both functional and structural abnormalities may improve when the obstructive process is relieved or removed. Trauma to the pancreas frequently results in duct injury and leakage, which may result in pseudocyst formation as well as local scar formation. Inadequately treated pancreatic trauma may result in persistent inflammatory changes in the distal gland.117
Pancreas divisum represents a special case of obstructive pancreatitis. It is the most common congenital anomaly involving the pancreas and occurs in up to 10% of children. It is thought to predispose the pancreas to recurrent acute pancreatitis and chronic pancreatitis, due to functional obstruction of a diminutive duct of Santorini that fails to communicate with Wirsung’s duct (Fig. 33-20). However, the classic picture of obstructive pancreatopathy with a dilated dorsal duct is unusual in pancreas divisum, so a decompressive operation or a lesser papilla sphincteroplasty is frequently not feasible or unsuccessful. Endoscopic stenting through the lesser papilla may result in temporary relief of symptoms, and this response would increase the possibility that a permanent surgical or endoscopic intervention will be successful. Although some authors emphasize the pathologic implications of pancreas divisum,118 others express skepticism that it represents a true risk to pancreatic secretory capacity or contributes to the development of chronic pancreatitis.119,120A recent French study reveals that pancreas divisum is equally prevalent among patients with idiopathic chronic pancreatitis and normal controls (7%), and is minimally increased in patients with PRSS1 and SPINK1 mutations. It is accompanied by CFTR mutations in 47% of patients, however, suggesting that the presence of pancreas divisum together with mutational events may increase the susceptibility to pancreatitis.121
Figure 33-20.
Pancreas divisum. Normal pancreatic duct anatomy and the variations of partial or complete pancreas divisum are shown. (Reproduced with permission of Wiley-Blackwell. From Warshaw.118)
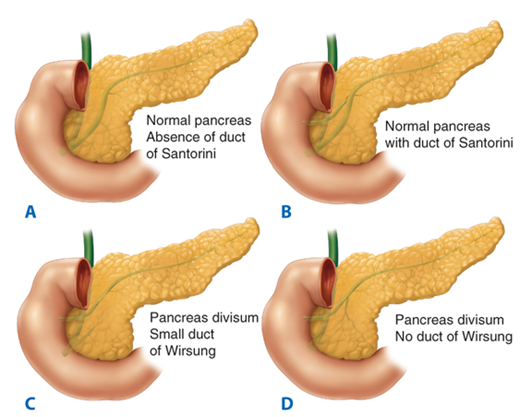
Chronic inflammatory pancreatitis is characterized by diffuse fibrosis and a loss of acinar elements with a predominant mononuclear cell infiltration throughout the gland.
A variant of chronic pancreatitis is a nonobstructive, diffusely infiltrative disease associated with fibrosis, a mononuclear cell (lymphocyte, plasma cell, or eosinophil) infiltrate, and an increased titer of one or more autoantibodies.122 This type, referred to as autoimmune pancreatitis (AIP), is associated with a variety of illnesses with suspected or proven autoimmune etiology, such as Sjögren’s syndrome, rheumatoid arthritis, and type I diabetes mellitus. AIP has been characterized as either type I, with accompanying systemic or multiorgan dysfunction, or type II, which is restricted to the pancreas.
Compressive stenosis of the intrapancreatic portion of the common bile duct is frequently seen in both types of AIP, along with symptoms of obstructive jaundice. Increased levels of serum β-globulin or immunoglobulin G4 are also present. Steroid therapy is uniformly successful in ameliorating the disease, including any associated bile duct compression.123 CFTR mutations which result in dislocation of the transmembrane protein have been found in AIP, and steroid therapy results in a normalization of the CFTR localization, and a resumption of normal chloride and bicarbonate secretion.124The differential diagnosis includes lymphoma, plasmacytoma (“pseudotumor” of the pancreas), and diffuse infiltrative carcinoma. Although the diagnosis is confirmed on pancreatic biopsy, presumptive treatment with steroids is usually undertaken, especially when clinical and laboratory findings, such as an elevation in IgG4 levels, support the diagnosis. Failure to obtain a cytologic specimen may lead to an unnecessary resectional procedure, and an untreated inflammatory component may cause sclerosis of the extrahepatic or intrahepatic bile ducts, with eventual liver failure.125
Chronic pancreatitis is highly prevalent among adolescents and young adults in Indonesia, southern India, and tropical Africa. Abdominal pain develops in adolescence, followed by the development of a brittle form of pancreatogenic diabetes. Parenchymal and intraductal calcifications are seen, and the pancreatic duct stones may be quite large.126 Many of the patients appear malnourished, some present with extreme emaciation, and a characteristic cyanotic coloration of the lips may be seen.127 In addition to protein-caloric malnutrition, toxic products of some indigenous foodstuffs have also been thought to contribute to the disease. Because of the geographic concentration of this early-onset form of chronic pancreatitis, it has been termed “tropical pancreatitis,” although the exact etiology remains unclear.
Clinically, tropical pancreatitis presents much like hereditary pancreatitis, and a familial pattern among cases is not unusual. SPINK1mutations have been documented in 20% to 55% of patients with tropical pancreatitis, and CFTR mutations have been reported as well.90,91The accelerated deterioration of endocrine and exocrine function, the chronic pain due to obstructive disease, and the recurrence of symptoms despite decompressive procedures characterize the course of disease. As immigrants from the tropical regions increasingly find their way to all parts of the world, an awareness of this severe form of chronic pancreatitis is helpful for those who treat patients with pancreatic disease.
Pancreatic fibrosis is seen in some asymptomatic elderly patients, in tropical populations, or in asymptomatic alcohol users. There is diffuse perilobar fibrosis and a loss of acinar cell mass, but without a main ductular component.
A shortcoming of these clinical classification systems is the lack of histologic criteria of chronic inflammation due to the usual absence of a biopsy specimen. The differentiation of recurrent acute pancreatitis from chronic pancreatitis with exacerbations of pain can be difficult to establish and is not facilitated by the current system. Similarly, cystic fibrosis is known to cause fibrosis and acinar dysfunction but is not included in the classification despite increasing evidence for its possible role in idiopathic chronic pancreatitis.128 Therefore, further refinements in the classification system for chronic pancreatitis are needed to allow a better prediction of its clinical course and a more accurate diagnosis of a likely etiologic agent.
When a definable cause for chronic pancreatitis is lacking, the term idiopathic is used to categorize the illness. Classically, the idiopathic group includes young adults and adolescents who lack a family history of pancreatitis but who may represent individuals with spontaneous gene mutations encoding regulatory proteins in the pancreas. A variable percentage of SPINK1 and CFTR mutations have been described in various studies. In addition, the idiopathic group has included a large number of older patients for whom no obvious cause of recurrent or chronic pancreatitis can be found.129 However, because the prevalence of biliary calculi increases steadily with age, it is not surprising that, as methods of biliary stone detection have improved, many elderly “idiopathic” pancreatitis patients are found to have biliary tract disease.130
As previously noted, an increasing number of mutations of the SPINK1 and CFTR genes have been identified in association with various forms of chronic pancreatitis. However, the role of genetic analysis in the management of these patients remains unclear, as guidelines have yet to be developed to allow physicians to use the data consistently.32 The clinical management of patients who harbor a minor CFTR mutation and chronic pancreatitis, for example, is still dictated by the clinical manifestations of the pancreatitis.
In early chronic pancreatitis, the histologic changes are unevenly distributed and are characterized by induration, nodular scarring, and lobular regions of fibrosis (Fig. 33-21). As the disease progresses, there is a loss of normal lobulation, with thicker sheets of fibrosis surrounding a reduced acinar cell mass, and dilatation of ductular structures (Fig. 33-22). The ductular epithelium is usually atypical and may display features of dysplasia, as evidenced by cuboidal cells with hyperplastic features, accompanied by areas of mononuclear cell infiltrates or patchy areas of necrosis. Cystic changes may be seen, but areas of relatively intact acinar elements and normal-appearing islets persist. In severe chronic pancreatitis, there is considerable replacement of acinar tissue by broad, coalescing areas of fibrosis, and the islet size and number are reduced (Fig. 33-23). Small arteries appear thickened and neural trunks become prominent.131
Figure 33-21.
Histology of early chronic pancreatitis. High-power microscopic (40x) histology of chronic pancreatitis shows an infiltration of mononuclear inflammatory cells throughout the interstitium of the pancreas, with little fibrosis. (Courtesy of Rhonda Yantiss, Weill Cornell Medical College.)
Figure 33-22.
Gross appearance of chronic pancreatitis. Areas of fibrosis and scarring are seen adjacent to other areas within the gland in which the lobar architecture is grossly preserved. A dilated pancreatic duct indicates the presence of downstream obstruction in this specimen removed from a patient with chronic pancreatitis. (Courtesy of Rhonda Yantiss, Weill Cornell Medical College.)

Figure 33-23.
Histology of severe chronic pancreatitis. High-power microscopic (40×) histologic appearance of advanced chronic pancreatitis shows extensive sheets of fibrosis and loss of acinar tissue, with preservation of islet tissue in scattered areas. (Courtesy of Rhonda Yantiss, Weill Cornell Medical College.)
Tropical pancreatitis and hereditary pancreatitis are histologically indistinguishable from chronic alcoholic pancreatitis. In obstructive chronic pancreatitis, calculi are absent, although periacinar fibrosis and dilated ductular structures are prominent. In pancreatic lobular fibrosis seen in elderly subjects, small ducts are dilated, sometimes with small calculi trapped within. Hypertrophy of ductular epithelia is thought to cause this small-duct disease, which is accompanied by perilobular fibrosis.132
A common feature of all forms of chronic pancreatitis is the perilobular fibrosis that forms surrounding individual acini, then propagates to surround small lobules, and eventually coalesces to replace larger areas of acinar tissue. The pathogenesis of this process involves the activation of pancreatic stellate cells (PSCs) that are found adjacent to acini and small arteries.133 The extended cytoplasmic processes of PSCs encircle the acini but appear quiescent in the normal gland, where they contain lipid vacuoles and cytoskeletal proteins. In response to pancreatic injury, the PSCs become activated and proliferate (similarly to hepatic stellate cells), lose their lipid vesicles, and transform into myofibroblast-like cells. These cells respond to proliferative factors such as transforming growth factor beta, platelet-derived growth factor, and proinflammatory cytokines and synthesize and secrete type I and III collagen and fibronectin. Studies indicate that vitamin A metabolites, similar to those present in quiescent PSCs, can inhibit the collagen production of activated cultured PSCs.134 This raises the possibility that early intervention may be possible to interrupt or prevent the fibrosis resulting from ongoing activation of PSCs.
The overall pathogenic sequence proposed by Schneider and Whitcomb135 whereby alcohol induces acute pancreatitis and, with ongoing exposure, promotes the development of chronic fibrosis, is summarized in Fig. 33-19. PSCs surrounding the acinus are activated in acute pancreatitis but may be inactivated by anti-inflammatory cytokines and, in the absence of further injury, may revert to a quiescent state. The role of proinflammatory macrophages, cytokines, and PSCs in models of acute and chronic pancreatitis represents an important area of current research.
Pancreatic stones are composed largely of calcium carbonate crystals trapped in a matrix of fibrillar and other material. The fibrillar center of most stones contains no calcium but rather a mixture of other metals. This suggests that stones form from an initial noncalcified protein precipitate, which serves as a focus for layered calcium carbonate precipitation. The same low-molecular-weight protein is present in stones and protein plugs and was initially named pancreatic stone protein, or PSP.136 PSP was found to be a potent inhibitor of calcium carbonate crystal growth and has subsequently been renamed lithostathine.137 Independently, a 15-kDa fibrillar protein isolated from the pancreas was named pancreatic thread protein, and it has been shown to be homologous with lithostathine. Finally, a protein product of the reg gene, so named because it is expressed in association with regenerating islets in models of pancreatic injury, was isolated and called reg protein, and was subsequently found to be homologous with lithostathine.138 No overall homology has been found between lithostathine and other pancreatic proteins. The PSP/pancreatic thread protein/reg/lithostathine gene encodes for a 166-amino acid product that undergoes posttranslational modification to produce isoforms present in pancreatic juice. The protein is expressed in all rodents and mammals, both in the pancreas as well as in brain tissue, where it is found in particularly high concentrations in pyramidal neurons in Alzheimer’s disease and Down syndrome. It is also found in the renal tubules, which is consistent with its biologic action of preventing calcium carbonate precipitation.
Calcium and bicarbonate ions are normally present in pancreatic juice in high concentrations, and the solubility product of calcium carbonate is greatly exceeded under normal conditions. Microcrystals of calcium carbonate can be seen in normal pancreatic juice but are usually clinically silent. Lithostathine is a potent inhibitor of calcium carbonate crystal formation, at a concentration of only 0.1 μmol/L. However, lithostathine concentrations in normal pancreatic juice are in the range of 20 to 25 μmol/L, so a constant suppression of calcium carbonate crystal formation is present in the normal pancreas.
In alcoholics and in patients with alcoholic chronic pancreatitis, lithostathine expression and secretion are dramatically inhibited139 (Fig. 33-24). In addition, elevated levels of precipitated lithostathine in the duct fluid in chronic pancreatitis patients suggests that the availability of the protein may be further reduced by the action of increased proteases and other proteins present in the duct fluid of alcoholic patients. Increased pancreatic juice protein levels in alcoholic men are reversible by abstinence from alcohol,140
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


