KEY POINTS
Modern cancer therapy is multidisciplinary, involving coordinated care by surgeons, medical oncologists, radiation oncologists, reconstructive surgeons, pathologists, radiologists, and primary care physicians.
Understanding cancer biology is essential to successfully implement personalized cancer therapy.
The following alterations are critical for malignant cancer growth: self-sufficiency of growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis, potential for limitless replication, angiogenesis, invasion and metastasis. Reprogramming of energy metabolism and evading immune destruction.
ONCOLOGY AND SURGICAL PRACTICE
As the population ages, oncology is becoming a larger portion of surgical practice. The surgeon often is responsible for the initial diagnosis and management of solid tumors. Knowledge of cancer epidemiology, etiology, staging, and natural history is required for initial patient assessment, as well as to determination of the optimal surgical therapy.
Modern cancer therapy is multidisciplinary, involving the coordinated care of patients by surgeons, medical oncologists, radiation oncologists, reconstructive surgeons, pathologists,radiologists, and primary care physicians. Primary (or definitive) surgical therapy refers to en bloc resection of tumor with adequate margins of normal tissues and regional lymph nodes as necessary. Adjuvant therapy refers to radiation therapy and systemic therapies, including chemotherapy, immunotherapy, hormonal therapy, and, increasingly, biologic therapy. The primary goal of surgical and radiation therapy is local and regional control. On the other hand, the primary goal of systemic therapy is systemic control by treatment of distant foci of subclinical disease to prevent distant recurrence. Surgeons must be familiar with adjuvant therapies to coordinate multidisciplinary care and to determine the best sequence of therapy.
Recent advances in molecular biology are revolutionizing medicine. New information is being translated rapidly into clinical use, with the development of new prognostic and predictive markers and new biologic therapies. Increasingly cancer therapy is getting personalized, incorporating information about each patient’s tumor characteristics, patient’s own genome, as well as host immune responses and tumor microenvironment, into clinical decision-making. It is therefore essential that surgeons understand the principles of molecular oncology to appropriately interpret these new contributions and incorporate them into practice.
EPIDEMIOLOGY
The term incidence refers to the number of new cases occurring. Incidence is usually expressed as the number of new cases per 100,000 persons per year. Mortality refers to the number of deaths occurring and is expressed as the number of deaths per 100,000 persons per year. Incidence and mortality data are usually available through cancer registries. Mortality data are also available as public records in many countries where deaths are registered as vital statistics, often with the cause of death. In areas where cancer registries do not exist, mortality data are used to extrapolate incidence rates. These numbers are likely to be less accurate than registry data, as the relationship between incidence and cause-specific death is likely to vary significantly among countries owing to the variation in health care delivery.
The incidence of cancer varies by geography. This is due in part to genetic differences and in part to differences in environmental and dietary exposures. Epidemiologic studies that monitor trends in cancer incidence and mortality have tremendously enhanced our understanding of the etiology of cancer. Furthermore, analysis of trends in cancer incidence and mortality allows us to monitor the effects of different preventive and screening measures, as well as the evolution of therapies for specific cancers.
The two types of epidemiologic studies that are conducted most often to investigate the etiology of cancer and the effect of prevention modalities are cohort studies and case-control studies. Cohort studies follow a group of people who initially do not have a disease over time and measure the rate of development of a disease. In cohort studies, a group that is exposed to a certain environmental factor or intervention usually is compared to a group that has not been exposed (e.g., smokers vs. nonsmokers). Case-control studies compare a group of patients affected with a disease to a group of individuals without the disease for a given exposure. The results are expressed in terms of an odds ratio, or relative risk. A relative risk <1 indicates a protective effect of the exposure, whereas a relative risk >1 indicates an increased risk of developing the disease with exposure.
In the year 2013, it is estimated that 1.6 million new cancer cases will be diagnosed in the United States, excluding carcinoma in situ of any site except bladder, and excluding basal cell and squamous cell carcinomas of the skin.1 In addition, 64,640 cases of carcinoma in situ of the breast, and 61,300 of melanoma in situ are expected.1
It is estimated that in 2013 estimated 580,350 people will die of cancer in the United States, corresponding to about 1600 deaths per day.1 The estimated new cancer cases and deaths by cancer type are shown in Table 10-1.1 The most common causes of cancer death in men are cancers of the lung and bronchus, prostate, and colon and rectum; in women, cancers are of the lung and bronchus, breast, and colon and rectum.1 These four cancers account for almost half (48%) of total cancer deaths among men and women.
ESTIMATED NEW CASES | ESTIMATED DEATHS | ESTIMATED NEW CASES | ESTIMATED DEATHS | ||
|---|---|---|---|---|---|
All cancers | 1,660,290 | 580,350 | Genital system | 339,810 | 58,480 |
Oral cavity and pharynx | 41,380 | 7,890 | Uterine cervix | 12,340 | 4,030 |
Digestive system | 290,200 | 144,570 | Uterine corpus | 49,560 | 8,190 |
Esophagus | 17,990 | 15,210 | Ovary | 22,240 | 14,030 |
Stomach | 21,600 | 10,990 | Vulva | 4,700 | 990 |
Small intestine | 8,810 | 1,170 | Vagina and other genital, female | 2,890 | 840 |
Colon and rectum | 142,820 | 50,830 | Prostate | 238,590 | 29,720 |
Anus, anal canal, and anorectum | 7,060 | 880 | Testis | 7,920 | 370 |
Liver and intrahepatic bile duct | 30,640 | 21,670 | Penis and other genital, male | 1,570 | 310 |
Gallbladder and other biliary | 10,310 | 3,230 | Urinary system | 140,430 | 29,790 |
Pancreas | 45,220 | 38,460 | Urinary bladder | 72,570 | 15,210 |
Other digestive organs | 5,750 | 2,130 | Kidney and renal pelvis | 65,150 | 13,680 |
Respiratory system | 246,210 | 163,890 | Ureter and other urinary organs | 2,710 | 900 |
Larynx | 12,260 | 3,630 | Eye and orbit | 2,800 | 320 |
Lung and bronchus | 228,190 | 159,480 | Brain and other nervous system | 23,130 | 14,080 |
Other respiratory organs | 5,760 | 780 | Endocrine system | 62,710 | 2,770 |
Bones and joints | 3,010 | 1,440 | Thyroid | 60,220 | 1,850 |
Soft tissue (including heart) | 11,410 | 4,390 | Other endocrine | 2,490 | 920 |
Skin (excluding basal and squamous) | 82,770 | 12,650 | Lymphoma | 79,030 | 20,200 |
| Melanoma | 76,690 | 9,480 | Multiple myeloma | 22,350 | 10,710 |
| Other nonepithelial | 6,080 | 3,170 | Leukemia | 48,610 | 23,720 |
| Breast | 234,580 | 40,030 | Other and unspecified primary sitesb | 31,860 | 45,420 |
The annual age-adjusted cancer incidence rates among males and females for selected cancer types are shown in Fig. 10-1.1 Incidence rates are declining for most cancer sites, but they are increasing among both men and women for melanoma of the skin, cancers of the liver and thyroid (Fig. 10-2).1 Incidence rates are decreasing for all four major cancer sites except for breast cancer in women. Age-adjusted incidence rate of breast cancer started to decrease from 2001 to 2004.2 This decrease in breast cancer incidence has at least temporally been associated with the first report of the Women’s Health Initiative, which documented an increased risk of coronary artery disease and breast cancer with the use of hormone replacement therapy; this was followed by a drop in the use of hormone replacement therapy by postmenopausal women in the United States.2 Unfortunately after this initial drop, breast cancer incidence has remained relatively stable from 2005 to 2009.
Figure 10-1.
Ten leading cancer types with the estimated new cancer cases and deaths by sex in the United States, 2013. *Excludes basal and squamous cell skin cancers and in situ carcinomas except those of the urinary bladder. Estimates are rounded to the nearest 10 (Modified with permission from John Wiley and Sons: Siegel R et al. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11. © 2013 American Cancer Society, Inc.)
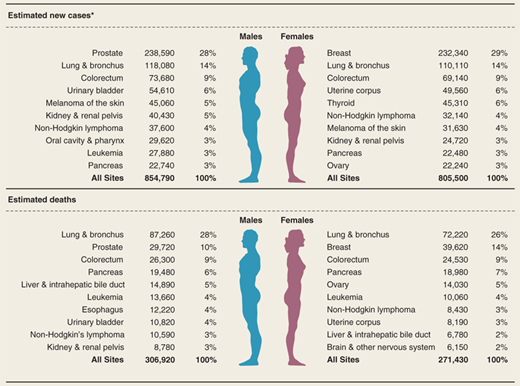
Figure 10-2.
Trends in cancer incidence rates for selected cancer by sex among males and females for selected cancer types, United States, 1975 to 2009. Rates are age adjusted to the 2000 U.S. standard population. (Modified with permission from John Wiley and Sons: Siegel R et al. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11. © 2013 American Cancer Society, Inc.)1 +Liver includes intrahepatic bile duct
Declines in colorectal cancer incidence have been mainly attributed to increased screening that allows for removal of precancerous polyps. Prostate cancer rates rapidly increased and decreased between 1995 and 1998. These trends are thought to be attributable to increased use of prostate-specific antigen (PSA) screening.3 Although analysis now suggest prostate cancer incidence has declined steadily by 1.9% per year from 2000 to 2009, annual rates fluctuate likely reflecting variations in screening.
Differences in lung cancer incidence patterns between women and men are thought to reflect historical differences in tobacco use. Differences in smoking prevalence is also thought to contribute to regional differences in lung cancer incidence. Lung cancer incidence is fourfold higher in Kentucky which has the highest smoking prevalence, compared with Utah, that has the lowest smoking prevalence (128 vs. 34 lung cancer cases per 100,000 men).1
The 5-year survival rates for selected cancers are listed in Table 10-2. From 2005 to 2009, cancer death rates decreased by 1.8% per year in males and by 1.5% per year in females.1 These declines in mortality have been consistent in the past decade, and larger than what was observed in the previous decade.3 Over the past two decades, death rates have decreased from their peak by more than 30% for colorectal cancer, female breast cancer, male lung cancer and more than 40% for prostate cancer. The decrease in lung cancer death rates in men is thought to be due to a decrease in tobacco use, whereas the decreases in death rates from breast, colorectal cancer, and prostate cancer reflect advances in early detection and treatment.
RELATIVE 5-YEAR SURVIVAL RATES (%) | |||
|---|---|---|---|
CANCER TYPE | 1975–1977 | 1987–1989 | 2002-2008 |
All cancers | 49 | 56 | 68 |
Brain | 22 | 29 | 35 |
Breast (female) | 75 | 84 | 90 |
Uterine cervix | 69 | 70 | 69 |
Colon | 51 | 61 | 65 |
Uterine corpus | 87 | 83 | 83 |
Esophagus | 5 | 10 | 19 |
Hodgkin’s disease | 72 | 79 | 87 |
Kidney | 50 | 57 | 72 |
Larynx | 66 | 66 | 63 |
Leukemia | 34 | 43 | 58 |
Liver | 3 | 5 | 16 |
Lung and bronchus | 12 | 13 | 17 |
Melanoma of the skin | 82 | 88 | 93 |
Multiple myeloma | 25 | 28 | 43 |
Non-Hodgkin’s lymphoma | 47 | 51 | 71 |
Oral cavity | 53 | 54 | 65 |
Ovary | 36 | 38 | 43 |
Pancreas | 2 | 4 | 6 |
Prostate | 68 | 83 | 100 |
Rectum | 48 | 58 | 68 |
Stomach | 15 | 20 | 28 |
Testis | 83 | 95 | 96 |
Thyroid | 92 | 95 | 98 |
Urinary bladder | 73 | 79 | 80 |
The five most common cancers for men worldwide are lung, prostate, colorectal cancer, stomach, liver, and for women are breast, colorectal, cervix, lung, and stomach.4 Notably, for several cancer types there is wide geographical variability in cancer incidence (Fig. 10-3). The mortality rates for different cancers also vary significantly among countries. This is attributable not only to variations in incidence but also to variations in survival after a cancer diagnosis. The survival rates are influenced by treatment patterns as well as by variations in cancer screening practices, which affect the stage of cancer at diagnosis. For example, the 5-year survival rate for stomach cancer is much higher in Japan, where the cancer incidence is high enough to warrant mass screening, which is presumed to lead to earlier diagnosis. In the case of prostate cancer, on the other hand, the mortality rates diverge much less than the incidence rates among countries. Survival rates for prostate cancer are much higher in North America than in developing countries.5 It is possible that the extensive screening practices in the United States allow discovery of cancers at an earlier, more curable stage; however, it is also possible that this screening leads to discovery of more latent, less biologically aggressive cancers, which may not have caused death even if they had not been identified.
Figure 10-3.
Estimated cancer incidence worldwide in 2008. Age-standardized incidence rates per 100,000 for all cancers (upper left), breast cancer (upper right), liver cancer (lower left), and stomach cancer (lower right). (Modified with permission from Ferlay, IARC)4

About one million new cases of stomach cancer were estimated to have occurred in 2008 (988,000 cases, 7.8% of the total), making it the fourth most common malignancy in the world, behind cancers of the lung, breast, and colorectal cancer. The incidence of stomach cancer varies significantly among different regions of the world. The difference in risk by country is presumed to be primarily due to differences in dietary factors. The risk is increased by high consumption of preserved salted foods such as meats and pickles, and decreased by high intake of fruits and vegetables.5 There also is some international variation in the incidence of infection with Helicobacter pylori, which is known to play a major role in gastric cancer development.5 Fortunately, a steady decline is being observed in the incidence and mortality rates of gastric cancer. This may be related to improvements in preservation and storage of foods as well as due to changes in the prevalence of H. pylori.5 More than 70% of cases (713,000 cases) occur in developing countries, and half the cases in the world occur in Eastern Asia (mainly in China).4 Age-standardized incidence rates are about twice as high for men as for women, ranging from 3.9 in Northern Africa to 42.4 in Eastern Asia for men, and from 2.2 in Southern Africa to 18.3 in Eastern Asia for women. Stomach cancer is the second leading cause of cancer death in both sexes worldwide.
Overall, the incidence of breast cancer is rising in most countries. Incidence varies from 19.3 per 100,000 women in Eastern Africa to 89.7 per 100,000 women in Western Europe, and are high in developed regions of the world (except Japan) and low in most of the developing regions.4 Although breast cancer has been linked to cancer susceptibility genes, mutations in these genes account for only 5% to 10% of breast tumors, which suggests that the wide geographic variations in breast cancer incidence are not due to geographic variations in the prevalence of these genes. Most of the differences, therefore, are attributed to differences in reproductive factors, diet, alcohol, obesity, physical activity, and other environmental differences. Indeed, breast cancer risk increases significantly in females who have migrated from Asia to America.5 The range of breast cancer mortality rates is much less (approximately 6 to 19 per 100,000) because of the more favorable survival of breast cancer in developed regions. As a result, breast cancer ranks as the fifth cause of death from cancer overall (458,000 deaths), but it is still the most frequent cause of cancer death in women in both developing (269,000 deaths, 12.7% of total) and developed regions (estimated 189,000 deaths). 4
There is a 25-fold variation in colon cancer incidence worldwide.5 The incidence of colon and rectal cancer is higher in developed countries than in developing countries. The incidence rates are highest in North America, Australia and New Zealand, and Western Europe, and especially in Japanese men.5 In contrast, the incidence is relatively low in North Africa, South America, and eastern, Southeastern, and Western Asia. These geographic differences are thought to reflect environmental exposures and are presumed to be related mainly to dietary differences in consumption of animal fat, meat, and fiber.5
Worldwide liver cancer is the fifth most common cancer in men (523,000 cases, 7.9% of the total) and the seventh in women (226,000 cases, 6.5% of the total). Almost 85% of liver cancer cases occur in developing countries, and particularly in men.4 The overall sex ratio male:female is 2:4. The regions of high incidence are Eastern and Southeastern Asia, Middle and Western Africa, as well as Melanesia and Micronesia/Polynesia (particularly in men). Low rates are estimated in developed regions, with the exception of Southern Europe. There were an estimated 694,000 deaths from liver cancer in 2008 (477,000 in men, 217,000 in women), and because of its high fatality (overall ratio of mortality to incidence of 0.93), liver cancer is the third most common cause of death from cancer worldwide. The geographical distribution of the mortality rates is similar to that observed for incidence. Worldwide, the major risk factors for liver cancer are infection with hepatitis B and C viruses and consumption of foods contaminated with aflatoxin. Hepatitis B immunization in children has recently been shown to reduce the incidence of liver cancer.5
In summary, the incidence rates of many common cancers vary widely by geography. This is due in part to genetic differences, including racial and ethnic differences. It is due also in part to differences in environmental and dietary exposures, factors that can potentially be altered. Therefore, establishment of regional and international databases is critical to improving our understanding of the etiology of cancer and will ultimately assist in the initiation of targeted strategies for global cancer prevention. Furthermore, the monitoring of cancer mortality rates and 5-year cancer-specific survival rates will identify regions where there are inequities of health care, so that access to health care can be facilitated and guidelines for treatment can be established.
CANCER BIOLOGY
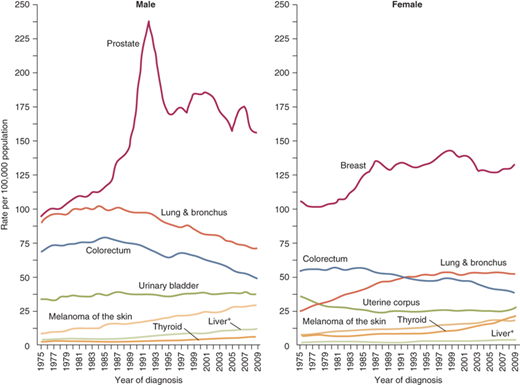
Although there are >100 types of cancer, it has been proposed that there are six essential alterations in cell physiology that dictate malignant growth: self-sufficiency of growth signals, insensitivity to growth-inhibitory signals, evasion of apoptosis (programmed cell death), potential for limitless replication, angiogenesis, and invasion and metastasis.6 Recently two additional hallmarks have emerged—reprogramming of energy metabolism and evading immune destruction.7 These hallmarks of cancer are being pursued as targets for cancer therapy (Figure 10-4).
Figure 10-4.
Hallmarks of cancer and their therapeutic implications.
Drugs that interfere with each of the acquired capabilities necessary for tumor growth and progression are in clinical trials and in some cases approved for clinical use in treating forms of human cancer. The drugs listed are illustrative examples.(Modified with permission from Hanahan et al. Copyright Elsevier.)7
In normal cells, cell growth and proliferation are under strict control. In cancer cells, cells become unresponsive to normal growth controls, which leads to uncontrolled growth and proliferation. Human cells require several genetic changes for neoplastic transformation. Cell type–specific differences also exist for tumorigenic transformation. Abnormally proliferating, transformed cells outgrow normal cells in the culture dish (i.e., in vitro) and commonly display several abnormal characteristics.8 These include loss of contact inhibition (i.e., cells continue to proliferate after a confluent monolayer is formed); an altered appearance and poor adherence to other cells or to the substratum; loss of anchorage dependence for growth; immortalization; and gain of tumorigenicity (i.e., the ability to give rise to tumors when injected into an appropriate host).
Tumorigenesis is proposed to have three steps: initiation, promotion, and progression. Initiating events such as gain of function of genes known as oncogenes or loss of function of genes known as tumor-suppressor genes may lead a single cell to acquire a distinct growth advantage. Although tumors usually arise from a single cell or clone, it is thought that sometimes not a single cell but rather a large number of cells in a target organ may have undergone the initiating genetic event. Thus, many normal-appearing cells may have an increased malignant potential. This is referred to as a field effect. The initiating events are usually genetic and occur as deletions of tumor-suppressor genes or amplification or mutation of oncogenes. Subsequent events can lead to accumulations of additional deleterious mutations in the clone.
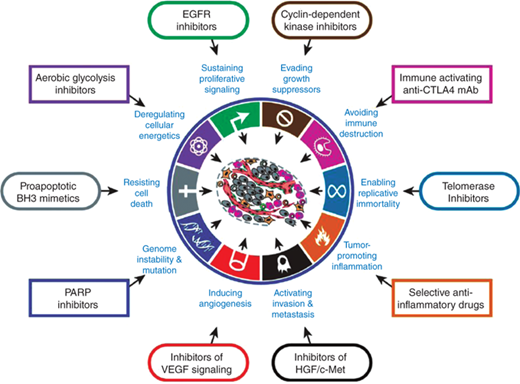
Cancer is thought to be a disease of clonal progression as tumors arise from a single cell and accumulate mutations that confer on the tumor an increasingly aggressive behavior. Most tumors go through a progression from benign lesions to in situ tumors to invasive cancers (e.g., atypical ductal hyperplasia to ductal carcinoma in situ to invasive ductal carcinoma of the breast). Fearon and Vogelstein proposed the model for colorectal tumorigenesis presented in Fig. 10-5.9 Colorectal tumors arise from the mutational activation of oncogenes coupled with mutational inactivation of tumor-suppressor genes, the latter being the predominant change.9 Mutations in at least four or five genes are required for formation of a malignant tumor, while fewer changes suffice for a benign tumor. Although genetic mutations often occur in a preferred sequence, a tumor’s biologic properties are determined by the total accumulation of its genetic changes.
Figure 10-5.
A genetic model for colorectal tumorigenesis. Tumorigenesis proceeds through a series of genetic alterations involving oncogenes and tumor-suppressor genes. In general, the three stages of adenomas represent tumors of increasing size, dysplasia, and villous content. Individuals with familial adenomatous polyposis (FAP) inherit a mutation on chromosome arm 5q. In tumors arising in individuals without polyposis, the same region may be lost or mutated at a relatively early stage of tumorigenesis. A ras gene mutation (usually K-ras) occurs in one cell of a pre-existing small adenoma which, through clonal expansion, produces a larger and more dysplastic tumor. The chromosome arms most frequently deleted include 5q, 17p, and 18q. Allelic deletions of chromosome arms 17p and 18q usually occur at a later stage of tumorigenesis than do deletions of chromosome arm 5q or ras gene mutations. The order of these changes varies, however, and accumulation of these changes, rather than their order of appearance, seems most important. Tumors continue to progress once carcinomas have formed, and the accumulated chromosomal alterations correlate with the ability of the carcinomas to metastasize and cause death. DCC = deleted in colorectal cancer gene. (Modified with permission from Fearon et al. Copyright Elsevier.)9
Gene expression is a multistep process that starts from transcription of a gene into messenger ribonucleic acid (mRNA) and then translation of this sequence into the functional protein. There are several controls at each level. In addition to alterations at the genome level (e.g., amplifications of a gene), alterations at the transcription level (e.g., methylation of the DNA leading to transcriptional silencing) or at the level of mRNA processing, mRNA stability, mRNA translation, or protein stability, all can alter the levels of critical proteins and thus contribute to tumorigenesis. Alternatively, changes in the genomic sequence can lead to a mutated product with altered function.
The proliferative advantage of tumor cells is a result of their ability to bypass quiescence. Cancer cells often show alterations in signal transduction pathways that lead to proliferation in response to external signals. Mutations or alterations in the expression of cell-cycle proteins, growth factors, growth factor receptors, intracellular signal transduction proteins, and nuclear transcription factors all can lead to disturbance of the basic regulatory mechanisms that control the cell cycle, allowing unregulated cell growth and proliferation.
The cell cycle is divided into four phases (Fig. 10-6).10 During the synthetic or S phase, the cell generates a single copy of its genetic material, whereas in the mitotic or M phase, the cellular components are partitioned between two daughter cells. The G1 and G2 phases represent gap phases during which the cells prepare themselves for completion of the S and M phases, respectively. When cells cease proliferation, they exit the cell cycle and enter the quiescent state referred to as G0. In human tumor cell-cycle regulators like INK4A, INK4B, and KIP1 are frequently mutated or altered in expression. These alterations underscore the importance of cell-cycle regulation in the prevention of human cancers.
Figure 10-6.
Schematic representation of the phases of the cell cycle. Mitogenic growth factors can drive a quiescent cell from G0 into the cell cycle. Once the cell cycle passes beyond the restriction point, mitogens are no longer required for progression into and through S phase. The DNA is replicated in S phase, and the chromosomes are condensed and segregated in mitosis. In early G1 phase, certain signals can drive a cell to exit the cell cycle and enter a quiescent phase. Cell-cycle checkpoints have been identified in G1, S, G2, and M phases. CDK = cyclin-dependent kinase. (Adapted from Kastan et al)10

Normal cellular genes that contribute to cancer when abnormal are called oncogenes. The normal counterpart of such a gene is referred to as a proto-oncogene. Oncogenes are usually designated by three-letter abbreviations, such as myc or ras. Oncogenes are further designated by the prefix “v-” for virus or “c-” for cell or chromosome, corresponding to the origin of the oncogene when it was first detected. Proto-oncogenes can be activated (show increased activity) or overexpressed (expressed at increased protein levels) by translocation (e.g., abl), promoter insertion (e.g., c-myc), mutation (e.g., ras), or amplification (e.g., HER2/neu). More than 100 oncogenes have been identified.
Oncogenes may be growth factors (e.g., platelet-derived growth factor), growth factor receptors (e.g., HER2), intracellular signal transduction molecules (e.g., ras), nuclear transcription factors (e.g., c-myc), or other molecules involved in the regulation of cell growth and proliferation. Growth factors are ubiquitous proteins that are produced and secreted by cells locally and that stimulate cell proliferation by binding specific cell-surface receptors on the same cells (autocrine stimulation) or on neighboring cells (paracrine stimulation). Persistent overexpression of growth factors can lead to uncontrolled autostimulation and neoplastic transformation. Alternatively, growth factor receptors can be aberrantly activated (turned on) through mutations or overexpressed (continually presenting cells with growth-stimulatory signals, even in the absence of growth factors), which leads cells to respond as if growth factor levels are altered. The growth-stimulating effect of growth factors and other mitogens is mediated through postreceptor signal transduction molecules. These molecules mediate the passage of growth signals from the outside to the inside of the cell and then to the cell nucleus, initiating the cell cycle and DNA transcription. Aberrant activation or expression of cell-signaling molecules, cell-cycle molecules, or transcription factors may play an important role in neoplastic transformation. Protein tyrosine kinases account for a large portion of known oncogenes. One of the best-studied oncogenes, HER2 is discussed as an example later.
HER2, also known as neu or c-erbB-2, is a member of the epidermal growth factor receptor (EGFR) family and is one of the best-characterized tyrosine kinases. Unlike other receptor tyrosine kinases, HER2/neu does not have a direct soluble ligand. It plays a key role in signaling, however, because it is the preferred partner in heterodimer formation with all the other EGFR family members (EGFR/c-erbB-1, HER2/c-erbB-3, and HER3/c-erbB-4), which bind at least 30 ligands, including epidermal growth factor (EGF), transforming growth factor α (TGFα), heparin-binding EGF-like growth factor, amphiregulin, and heregulin.11 Heterodimerization with HER2 potentiates recycling of receptors rather than degradation, enhances signal potency and duration, increases affinity for ligands, and increases catalytic activity.11
HER2 can interact with different members of the HER family and activate mitogenic and antiapoptotic pathways (Fig. 10-7). The specificity and potency of the intracellular signals are affected by the identity of the ligand, the composition of the receptors, and the phosphotyrosine-binding proteins associated with the erbB molecules. The Ras- and Shc-activated mitogen-activated protein kinase (MAPK) pathway is a target of all erbB ligands, which increase the transcriptional activity of early response genes such as c-myc, c-fos, and c-jun.12 MAPK-independent pathways such as the phosphoinositide-3 kinase (PI3K) pathway also are activated by most erbB dimers, although the potency and kinetics of activation may differ. Stimulation of the PI3K pathway through HER2 signaling also can lead to activation of survival molecule Akt, which suppresses apoptosis through multiple mechanisms. The critical role of HER2 in cancer biology has been leveraged for therapeutics, leading to several HER2- targeted drugs with different mechanism of action approved by the Food and Drug Administration (FDA): monoclonal antibodies trastuzumab and pertuzumab, small molecule inhibitor lapatinib, and antibody-drug conjugate ado-trastuzumab emtansine.
Figure 10-7.
The HER2 signaling pathway. HER2 can interact with different members of the HER family and activate mitogenic and antiapoptotic pathways. 4E-BP1= eIF4E binding protein 1; CREB = cyclic adenosine monophosphate element binding; eIF4E = eukaryotic initiation factor 4E; EZH = enhancer of zeste homolog; FAK = focal adhesion kinase; Fas-L = Fas ligand; GSK3 = glycogen synthase kinase-3; HER = human epidermal growth receptor; IKK = IκB kinase; ILK= integrin-linked kinase; IP3 = inositol triphosphate; IκB = inhibitor of NF-κB; MAPK = mitogen-activated protein kinase; MDM2 = mouse double minute 2 homologue; MEK = mitogen-activated protein/extracellular signal regulated kinase kinase; MEKK = MEK kinase; mTOR = mammalian target of rapamycin; NF-κB = nuclear factor κB; PI3K = phosphoinositide-3 kinase; PLC-γ = phospholipase Cγ; SAPK = stress-activated protein kinase; SEK = SAPK/extracellular signal regulated kinase kinase; TSC = tuberous sclerosis complex. (Modified with permission from Meric-Bernstam et al.)171
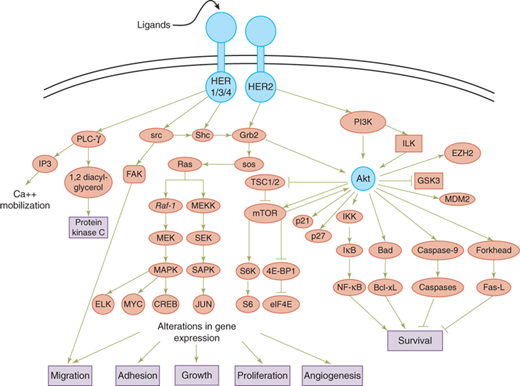
The mutant rat neu gene was first recognized as an oncogene in neuroblastomas from carcinogen-treated rats.13 The HER2 gene is frequently amplified and the protein overexpressed in many cancers, including breast, ovarian, lung, gastric, and oral cancers. Overexpression of HER2 results in ligand-independent activation of HER2 kinase, which leads to mitogenic signaling. HER2 overexpression is associated with increased cell proliferation and anchorage-independent growth as well as resistance to proapoptotic stimuli. Further, overexpression of HER2 increases cell migration and upregulates the activities of matrix metalloproteinases (MMPs) and in vitro invasiveness. In animal models, HER2 increases tumorigenicity, angiogenesis, and metastasis. These results all suggest that HER2 plays a key role in cancer biology. More recently HER2 mutations have also been reported in human cancer. HER2 mutations have been detected in 2% to 4% of nonsmall cell lung cancer.14,15,16,17 In frame insertions within exon 20 has been the most commonly reported mutation. HER2 mutations are more common in nonsmokers and are nonoverlapping with other oncogenic mutations in lung cancer (e.g., EGFR and Ras). Data from 8 breast cancer genome-sequencing projects identified 25 patients with HER2 somatic mutations in cancers lacking HER2 gene amplification.18 Seven of 13 mutations were functionally characterized and found to be activating mutations. All of these mutations were sensitive to the irreversible kinase inhibitor, neratinib. A prospective, multi-institutional clinical trial has been launched to screen patients with stage IV breast cancer for HER2 somatic mutations and determine the clinical outcome of treating them with HER2-targeted therapy.
Apoptosis is a genetically regulated program to dispose of cells. Cancer cells must avoid apoptosis if tumors are to arise. The growth of a tumor mass is dependent not only on an increase in proliferation of tumor cells but also on a decrease in their apoptotic rate. Apoptosis is distinguished from necrosis because it leads to several characteristic changes. In early apoptosis, the changes in membrane composition lead to extracellular exposure of phosphatidylserine residues, which avidly bind annexin, a characteristic that is used to discriminate apoptotic cells in laboratory studies. Late in apoptosis there are characteristic changes in nuclear morphology, such as chromatin condensation, nuclear fragmentation, and DNA laddering, as well as membrane blebbing. Apoptotic cells are then engulfed and degraded by phagocytic cells. The effectors of apoptosis are a family of proteases called caspases (cysteine-dependent and aspartate-directed proteases). The initiator caspases (e.g., 8, 9, and 10), which are upstream, cleave the downstream executioner caspases (e.g., 3, 6, and 7) that carry out the destructive functions of apoptosis.
Two principal molecular pathways signal apoptosis by cleaving the initiator caspases with the potential for crosstalk: the mitochondrial pathway and the death receptor pathway. In the mitochondrial (or intrinsic) pathway, death results from the release of cytochrome c from the mitochondria. Cytochrome c, procaspase 9, and apoptotic protease activating factor 1 (Apaf-1) form an enzyme complex, referred to as the apoptosome, that activates the effector caspases. In addition to these proteins, the mitochondria contain other proapoptotic proteins such as second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO. The mitochondrial pathway can be stimulated by many factors, including DNA damage, reactive oxygen species, or the withdrawal of survival factors. The permeability of the mitochondrial membrane determines whether the apoptotic pathway will proceed. The Bcl-2 family of regulatory proteins includes both proapoptotic proteins (e.g., Bax, BAD, and Bak) and antiapoptotic proteins (e.g., Bcl-2 and Bcl-xL). The activity of the Bcl-2 proteins is centered on the mitochondria, where they regulate membrane permeability. Growth factors promote survival signaling through the PI3K/Akt pathway, which phosphorylates and inactivates proapoptotic BAD. In contrast, growth factor withdrawal may promote apoptosis through signaling by unphosphorylated BAD. The heat shock proteins, including Hsp70 and Hsp27, are also involved in inhibition of downstream apoptotic pathways by blocking formation of the apoptosome complex and inhibiting release of cytochrome c from the mitochondria.19
The second principal apoptotic pathway is the death receptor pathway, sometimes referred to as the extrinsic pathway. Cell-surface death receptors include Fas/APO1/CD95, tumor necrosis factor receptor 1, and KILL-ER/DR5, which bind their ligands Fas-L, tumor necrosis factor (TNF), and TNF-related apoptosis-inducing ligand (TRAIL), respectively. When the receptors are bound by their ligands, they form a death-inducing signaling complex (DISC). At the DISC, procaspase 8 and procaspase 10 are cleaved, yielding active initiator caspases.20 The death receptor pathway may be regulated at the cell surface by the expression of “decoy” receptors for Fas (DcR3) and TRAIL (TRID and TRUNDD). The decoy receptors are closely related to the death receptors but lack a functional death domain; therefore, they bind death ligands but do not transmit a death signal. Another regulatory group is the FADD-like interleukin-1 protease-inhibitory proteins (FLIPs). FLIPs have homology to caspase 8; they bind to the DISC and inhibit the activation of caspase 8. Finally, inhibitors of apoptosis proteins (IAPs) block caspase 3 activation and have the ability to regulate both the death receptor and the mitochondrial pathway.
In human cancers, aberrations in the apoptotic program include increased expression of Fas and TRAIL decoy receptors; increased expression of antiapoptotic Bcl-2; increased expression of the IAP-related protein survivin; increased expression of c-FLIP; mutations or downregulation of proapoptotic Bax, caspase 8, APAF1, XAF1, and death receptors CD95, TRAIL-R1, and TRAIL-R2; alterations of the p53 pathway; overexpression of growth factors and growth factor receptors; and activation of the PI3K/Akt survival pathway.20
Autophagy (self-eating) is a major cellular pathway for protein and organelle turnover. This process helps maintain a balance between anabolism and catabolism for normal cell growth and development. Inability to activate autophagy in response to nutrient deprivation, or constitutive activation of autophagy in response to stress, can lead to cell death; thus autophagy is sometimes referred to as a second form of programmed cell death. Autophagy plays an essential role during starvation, cellular differentiation, cell death, and aging. Autophagy is also involved in the elimination of cancer cells by triggering a nonapoptotic cell death program, which suggests a negative role in tumor development. Mouse models that are heterozygotes for the beclin 1 gene, an important gene for autophagy, have altered autophagic response and show a high incidence of spontaneous tumors, which establishes a role for autophagy in tumor suppression.21 This also suggests that mutations in other genes operating in this pathway may contribute to tumor formation through deregulation of autophagy. However, autophagy also acts as a stress response mechanism to protect cancer cells from low nutrient supply or therapeutic insults. Studies on the molecular determinants of autophagy are ongoing to determine whether autophagy can be modulated for therapeutic purposes.
A feature of malignant cells is their ability to invade the surrounding normal tissue. Tumors in which the malignant cells appear to lie exclusively above the basement membrane are referred to as in situ cancer, whereas tumors in which the malignant cells are demonstrated to breach the basement membrane, penetrating into surrounding stroma, are termed invasive cancer. The ability to invade involves changes in adhesion, initiation of motility, and proteolysis of the extracellular matrix (ECM).
Cell-to-cell adhesion in normal cells involves interactions between cell-surface proteins. Calcium adhesion molecules of the cadherin family (E-cadherin, P-cadherin, and N-cadherin) are thought to enhance the cells’ ability to bind to one another and suppress invasion. Migration occurs when cancer cells penetrate and attach to the basal matrix of the tissue being invaded; this allows the cancer cell to pull itself forward within the tissue. Attachment to glycoproteins of the ECM such as fibronectin, laminin, and collagen is mediated by tumor cell integrin receptors. Integrins are a family of glycoproteins that form heterodimeric receptors for ECM molecules. The integrins can form at least 25 distinct pairings of their α and β subunits, and each pairing is specific for a unique set of ligands. In addition to regulating cell adhesion to the ECM, integrins relay molecular signals regarding the cellular environment that influence shape, survival, proliferation, gene transcription, and migration.
Factors that are thought to play a role in cancer cell motility include autocrine motility factor, autotaxin, scatter factor (also known as hepatocyte growth factor), TGFα, EGF, and insulin-like growth factors.
Serine, cysteine, and aspartic proteinases and MMPs have all been implicated in cancer invasion. Urokinase and tissue plasminogen activators (uPA and tPA) are serine proteases that convert plasminogen into plasmin. Plasmin, in return, can degrade several ECM components. Plasmin also may activate MMPs. uPA has been more closely correlated with tissue invasion and metastasis than tPA. Plasminogen activator inhibitors 1 and 2 (PAI-1 and PAI-2) are produced in tissues and counteract the activity of plasminogen activators.
MMPs comprise a family of metal-dependent endopeptidases. Upon activation, MMPs degrade a variety of ECM components. Although MMPs often are referred to by their common names, which reflect the ECM component for which they have specificity, a sequential numbering system has been adopted for standardization. For example, collagenase-1 is now referred to as MMP-1. The MMPs are further classified as secreted and membrane-type MMPs. Most of the MMPs are synthesized as inactive zymogens (pro-MMP) and are activated by proteolytic removal of the propeptide domain outside the cell by other active MMPs or serine proteinases.
MMPs are upregulated in almost every type of cancer. Some of the MMPs are expressed by cancer cells, whereas others are expressed by the tumor stromal cells. Experimental models have demonstrated that MMPs promote cancer progression by increasing cancer cell growth, migration, invasion, angiogenesis, and metastasis. MMPs exert these effects by cleaving not only structural components of the ECM but also growth factor–binding proteins, growth factor precursors, cell adhesion molecules, and other proteinases. The activity of MMPs is regulated by their endogenous inhibitors and tissue inhibitors of MMPs (TIMP-1, TIMP-2, TIMP-3, and TIMP-4).
Angiogenesis is the establishment of new blood vessels from a pre-existing vascular bed. This neovascularization is essential for tumor growth and metastasis. Tumors develop an angiogenic phenotype as a result of accumulated genetic alterations and in response to local selection pressures such as hypoxia. Many of the common oncogenes and tumor-suppressor genes have been shown to play a role in inducing angiogenesis.
In response to the angiogenic switch, pericytes retract and the endothelium secretes several growth factors such as basic fibroblast growth factor, platelet-derived growth factor (PDGF), and insulin-like growth factor. The basement membrane and stroma around the capillary are proteolytically degraded, a process that is mediated in most part by uPA. The endothelium then migrates through the degraded matrix, initially as a solid cord and later forming lumina. Finally, sprouting tips anastomose to form a vascular network surrounded by a basement membrane.
Angiogenesis is mediated by factors produced by various cells, including tumor cells, endothelial cells, stromal cells, and inflammatory cells. The first proangiogenic factor was identified by Folkman and colleagues in 1971.22 Since then, several other factors have been shown to be proangiogenic or antiangiogenic. Of the angiogenic stimulators, the best studied are the vascular endothelial growth factors (VEGFs). The VEGF family consists of six growth factors (VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor) and three receptors (VEGFR1 or Flt-1, VEGFR2 or KDR/FLK-1, and VEGFR3 or Flt-4).23 Neuropilin 1 and 2 also may act as receptors for VEGF.24 VEGF is induced by hypoxia and by different growth factors and cytokines, including EGF, PDGF, TNF-α, TGFβ, and interleukin-1β. VEGF has various functions, including increasing vascular permeability, inducing endothelial cell proliferation and tube formation, and inducing endothelial cell synthesis of proteolytic enzymes such as uPA, PAI-1, urokinase plasminogen activator receptor, and MMP-1. Furthermore, VEGF may mediate blood flow by its effects on the vasodilator nitric oxide and act as an endothelial survival factor, thus protecting the integrity of the vasculature. The proliferation of new lymphatic vessels, lymphangiogenesis, is also thought to be controlled by the VEGF family. Signaling in lymphatic cells is thought to be modulated by VEGFR3.25 Experimental studies with VEGF-C and VEGF-D have shown that they can induce tumor lymphangiogenesis and direct metastasis via the lymphatic vessels and lymph nodes.25,26
PDGFs A, B, C, and D also play important roles in angiogenesis. PDGFs cannot only enhance endothelial cell proliferation directly but also upregulate VEGF expression in vascular smooth muscle cells, promoting endothelial cell survival via a paracrine effect.23 The angiopoietins angiopoietin-1 and angiopoietin-2 (Ang-1 and Ang-2), in return, are thought to regulate blood vessel maturation. Ang-1 and Ang-2 both bind angiopoietin-1 receptor (also known as tyrosine-protein kinase receptor TIE-2), but only the binding of Ang-1 activates signal transduction; thus Ang-2 is an Ang-1 antagonist. Ang-1, via the Tie-2 receptor, induces remodeling and stabilization of blood vessels. Upregulation of Ang-2 by hypoxic induction of VEGF inhibits Ang-1–induced Tie-2 signaling, which results in destabilization of vessels and makes endothelial cells responsive to angiogenic signals, thus promoting angiogenesis in the presence of VEGF. Therefore the balance between these factors determines the angiogenetic capacity of a tumor.
Tumor angiogenesis is regulated by several factors in a coordinated fashion. In addition to upregulation of proangiogenic molecules, angiogenesis also can be encouraged by suppression of naturally occurring inhibitors. Such inhibitors of angiogenesis include thrombospondin 1 and angiostatin. Angiogenesis is a prerequisite not only for primary tumor growth but also for metastasis. Angiogenesis in the primary tumor, as determined by microvessel density, has been demonstrated to be an independent predictor of distant metastatic disease and survival in several cancers. Expression of angiogenic factors such as VEGFs has had prognostic value in many studies. These findings further emphasize the importance of angiogenesis in cancer biology.
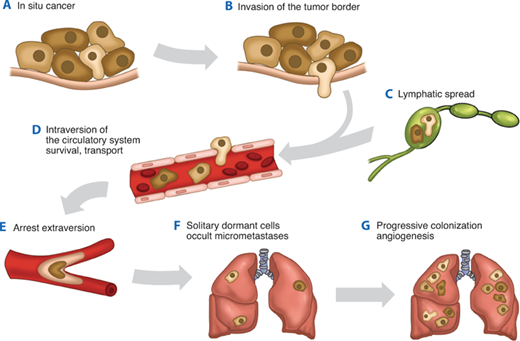
Metastases arise from the spread of cancer cells from the primary site and the formation of new tumors in distant sites. The metastatic process consists of a series of steps that need to be completed successfully (Fig. 10-8).27 First, the primary cancer must develop access to the circulation through either the blood circulatory system or the lymphatic system. After the cancer cells are shed into the circulation, they must survive. Next, the circulating cells lodge in a new organ and extravasate into the new tissue. Next, the cells need to initiate growth in the new tissue and eventually establish vascularization to sustain the new tumor. Overall, metastasis is an inefficient process, although the initial steps of hematogenous metastasis (the arrest of tumor cells in the organ and extravasation) are believed to be performed efficiently. Only a small subset of cancer cells is then able to initiate micrometastases, and an even smaller portion goes on to grow into macrometastases.
Figure 10-8.
A schematic representation of the metastatic process. A. The metastatic process begins with an in situ cancer surrounded by an intact basement membrane. B. Invasion requires reversible changes in cell-cell and cell–extracellular matrix adherence, destruction of proteins in the matrix and stroma, and motility. C. Metastasizing cells can enter the circulation via the lymphatics. D. They can also directly enter the circulation. E. Intravascular survival of the tumor cells and extravasation of the circulatory system follow. F. Metastatic single cells can colonize sites and remain dormant for years as occult micrometastases. G. Subsequent progression and neovascularization leads to clinically detectable metastases and progressively growing, angiogenic metastases. (Adapted by permission from Macmillan Publishers Ltd. Steeg PS. Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer. 2003;3:55. Copyright © 2003.)27
Metastases can sometimes arise several years after the treatment of primary tumors. For example, although most breast cancer recurrences occur within the first 10 years after the initial treatment and recurrences are rare after 20 years, breast cancer recurrences have been reported decades after the original tumor. This phenomenon is referred to as dormancy, and it remains one of the biggest challenges in cancer biology. Persistence of solitary cancer cells in a secondary site such as the liver or bone marrow is one possible contributor to dormancy.28 Another explanation of dormancy is that cells remain viable in a quiescent state and then become reactivated by a physiologically perturbing event. Interestingly, primary tumor removal has been proposed to be a potentially perturbing factor.29 An alternate explanation is that cells establish preangiogenic metastases in which they continue to proliferate but that the proliferative rate is balanced by the apoptotic rate. Therefore, when these small metastases acquire the ability to become vascularized, substantial tumor growth can be achieved at the metastatic site, leading to clinical detection.
Several types of tumors metastasize in an organ-specific pattern. One explanation for this is mechanical and is based on the different circulatory drainage patterns of the tumors. When different tumor types and their preferred metastasis sites were compared, 66% of organ-specific metastases were explained on the basis of blood flow alone. The other explanation for preferential metastasis is what is referred to as the “seed and soil” theory, the dependence of the seed (the cancer cell) on the soil (the secondary organ). According to this theory, once cells have reached a secondary organ, their growth efficiency in that organ is based on the compatibility of the cancer cell’s biology with its new microenvironment. For example, breast cancer cells may grow more efficiently in bone than in some other organs because of favorable molecular interactions that occur in the bone microenvironment. The ability of cancer cells to grow in a specific site likely depends on features inherent to the cancer cell, features inherent to the organ, and the interplay between the cancer cell and its microenvironment.30
Many of the oncogenes discovered to date, such as HER2 and ras, are thought to potentiate not only malignant transformation but also one or more of the steps required in the metastatic process. Experimental models have suggested a role for several molecules, including RhoC, osteopontin and interleukin-11, and Twist, in tumor metastasis. Metastasis also may involve the loss of metastasis-suppressor genes. Laboratory work involving cancer cell lines that have been selected to have a higher metastatic potential have led to the realization that these more highly metastatic cells have a different gene expression profile than their less metastatic parental counterparts. This in turn has led to the currently held belief that the ability of a primary tumor to metastasize may be predictable by analysis of its gene expression profile. Indeed, several studies have recently focused on identifying a gene expression profile or a molecular signature that is associated with metastasis. It has been shown that such a gene expression profile can be used to predict the probability that the patient will remain free of distant metastasis.31 This suggests that the metastatic potential of a tumor is already predetermined by the genetic alterations that the cancer cells acquire early in tumorigenesis. Notably, this hypothesis differs from the multistep tumorigenesis theory in that the ability to metastasize is considered an inherent quality of the tumor from the beginning. It is assumed that metastasis develops not from a few rare cells in the primary tumor that acquire the ability to metastasize but that all cells in tumors with such molecular signatures develop the ability to metastasize. The reality probably lies in between since some early genetic changes detectable in the entire tumor can give tumors an advantage in the metastatic process, whereas additional genetic changes can give a clone of cells additional advantages, thus allowing them to succeed in metastasis.
A regulatory program referred to as epithelial-mesenchymal transition (EMT) is a fundamental event in morphogenesis. During EMT epithelial cells are converted to migratory and invasive cells.32 EMT, has also been implicated as the mechanism through which epithelial cells acquire the ability to migrate, invade, resist apoptosis and metastasize. EMT is a developmental process, and a set of pleiotropically acting transcriptional factors, including Snail, Twist, Slug, and Zeb1/2orchestrateEMT. Several of these transcription factors can directly repress E-cadherin gene expression, depriving cancer cells of this key suppressor of motility and invasiveness. It has been proposed that the process of invasion and metastases requires significant plasticity, suggesting that EMT is required for invasion, intravasation and extravasation, and suppression of EMT regulators (and consequently EMT reversion, or MET) is required for metastatic outgrowth.33,34,35
Stem cells are cells that have the ability to perpetuate themselves through self-renewal and to generate mature cells of a particular tissue through differentiation.36 It has recently been proposed that stem cells themselves may be the target of transformation. It was first documented for leukemia and multiple myeloma that only a small subset of cancer cells is capable of extensive proliferation. It has subsequently also been shown for many solid cancers that only a small proportion of cells is clonogenic in culture and in vivo. In leukemia and multiple myeloma only a small subset of cancer cells is capable of extensive proliferation. Similarly, in many solid tumor types only a small proportion of cells is clonogenic in culture and in vivo. If indeed tumor growth and metastasis are driven by a small population of cancer stem cells, this may alter our current approaches to cancer therapy. Currently available drugs can shrink metastatic tumors but often cannot eradicate them. The failure of these treatments usually is attributed to the acquisition of drug resistance by the cancer cells; however, the cancer stem cell hypothesis raises the possibility that existing therapies may simply fail to kill cancer stem cells effectively. Therapeutic approaches targeting stem cells specifically are under study.
CANCER ETIOLOGY
One widely held opinion is that cancer is a genetic disease that arises from an accumulation of genomic alterations that leads to the selection of cells with increasingly aggressive behavior. These alterations may lead either to a gain of function by oncogenes or to a loss of function by tumor-suppressor genes. These acquired gene alterations are termed somatic mutations to distinguish them from germline mutations that are inherited from parents and transmitted to offspring. Somatic mutations in a cancer genome may consist of several classes of DNA sequence changes. These include substitutions of one base by another; insertions or deletions of small or large segments of DNA; rearrangements, in which the DNA sequence has been broken and then rejoined to another DNA segment; copy number losses that may result in complete absence of a DNA sequence and copy number gains from the two copies present in the normal diploid genome.
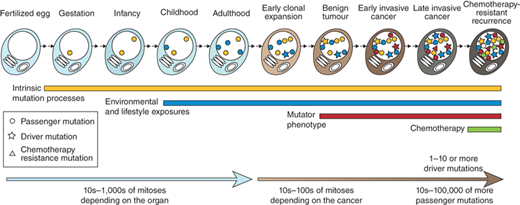
Somatic mutations in a cancer cell genome have accumulated over the lifetime of the patient (Fig. 10-9).37 DNA in normal cells is continuously damaged by internal and external mutagens. Most of this damage is repaired; however, a small fraction may remain as fixed mutations. Mutation rates increase in the presence of substantial exogenous mutagenic exposures, such as tobacco carcinogens or various forms of radiation, including ultraviolet light. These exposures are associated with increased rates of lung and skin cancer, respectively, and somatic mutations within such cancers often exhibit the distinctive mutational signatures known to be associated with the mutagen.38 The rates of somatic mutations are also increased in several rare inherited diseases, such as Fanconi anemia, ataxia telangiectasia, and xeroderma pigmentosum, which are associated with increased risks of cancer.39,40 The rest of the somatic mutations in a cancer cell have been acquired after the cancer cell already shows phenotypic evidence of neoplastic change. Whether the somatic mutation rate is always higher during this part of the lineage is controversial. This is clearly the case for some cancers. For instance, colorectal and endometrial cancers with defective DNA mismatch repair due to abnormalities in genes such as MLH1 and MSH2, exhibit increased rates of single nucleotide changes and small insertions/deletions atpolynucleotide tract.41 These tumor types are often referred to as “mutator phenotypes.”
Figure 10-9.
Accumulation of somatic mutations acquired by the cancer cell. Mutations may be acquired while the cell lineage is phenotypically normal, reflecting intrinsic mutations acquired during normal cell division as well as the effects of exogenous mutagens. Other processes such as example DNA repair defects may contribute to the mutational burden. Passenger mutations do not have any effect on the cancer cell, but driver mutations cause clonal expansion. Relapse after chemotherapy can be associated with resistance mutations that may predate the initiation of treatment.(Adapted by permission from Macmillan Publishers Ltd. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719. Copyright © 2009.)37
To date about 300 genes that have been reported to be mutated and causally implicated in cancer development.42 Ninety percent of cancer genes show somatic mutations in cancer, 20% show germline mutations, and 10% show both. The most common class of genomic alterations among the known cancer genes is a chromosomaltranslocation that creates a chimeric gene. Many more cancer genes have been found in leukemias, lymphomas, and sarcomas than in other types of cancer; and these genes are usually altered by chromosomal translocation. The most common cancer genes are protein kinases. Several domains that are involved in DNA binding and transcriptional regulation are also common in proteins encoded by cancer genes. Somatic mutations in a cancer genome may be classified according to its consequences for cancer development. “Driver” mutations confer a growth advantage to the cells carrying them and have been positively selected during the evolution of the cancer. The remainder of mutations are “bystanders” or “passengers” that do not confer growth advantage. It is likely that most somatic mutations are passenger mutations. Each tumor may have dozens to hundreds of genomic alterations, making it critical to determine which alterations are indeed drivers, and potentially better therapeutic targets.
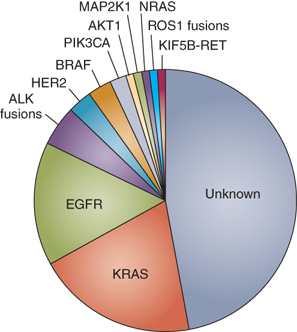
There are several ongoing large scale studies to characterize and catalogue genomic alterations in different cancer types, including the Cancer Genome Project at the Sanger Institute, United Kingdom, and The Cancer Genome Atlas project (TCGA). There are also increasing number of publically accessible resources, including COSMIC (http://www.sanger.ac.uk/cosmic), which curates comprehensive information on somatic mutations in human cancer.43 These resources are being utilized to determine the most common genomic alterations in common tumor types. This information is being integrated into clinical practice in many tumor types, such as lung cancer, where molecular drivers are being chosen taking into consideration in systemic therapy selection (Fig. 10-10).
Figure 10-10.
Molecular subsets of lung adenocarcinoma. Pie chart shows the percentage of tumors with each potentially actionable alteration.(Adapted by permission from Macmillan Publishers Ltd. Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349. Copyright © 2012.)172
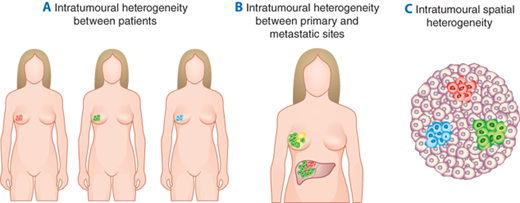
There is increasing recognition that tumors are heterogeneous; this represents an important challenge to utilizing genomic alterations to personalize cancer therapy (Fig. 10-11).44 First, there is significant intertumoral heterogeneity, such that patients with tumors that seem similar histologically, may differ in genomic alterations and in malignant potential.45,46,47 Second, during cancer progression, subclones frequently arise, resulting in differences in the proportion and pattern of genomic alterations between the primary tumor and the metastases or local-regional recurrences.44 Third, there may also be significant intratumoral heterogeneity, with spatially separated heterogeneous somatic mutations and chromosomal imbalances.48 Such spatial heterogeneity of subclones within the primary tumor or metastases provides an additional challenge, as it has been proposed that sequencing of a biopsy specimen or only a portion of the tumor could miss therapeutically relevant genomic alterations. The genomic alterations found in a tumor can also change under the selective pressure of a targeted therapy, adding to the challenge of implementing genomically-informed personalized therapy.
Figure 10-11.
“Two-hit” tumor formation in both hereditary and nonhereditary cancers. A “one-hit” clone is a precursor to the tumor in nonhereditary cancer, whereas all cells are one-hit clones in hereditary cancer. (Adapted by permission from Macmillan Publishers Ltd. Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157. Copyright © 2001.)51
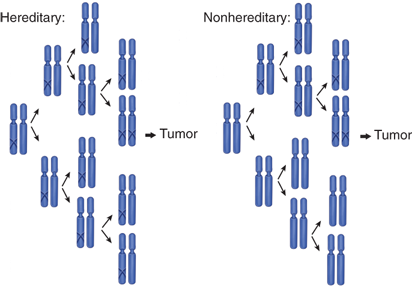
Most of our information on human cancer genes has been gained from hereditary cancers. In the case of hereditary cancers, the individual carries a particular germline mutation in every cell. To date, over 70 genes have been associated with hereditary cancers (Table 10-3).42 A few of these hereditary cancer genes are oncogenes, but most are tumor-suppressor genes. Although hereditary cancer syndromes are rare, somatic mutations that occur in sporadic cancer have been found to disrupt the cellular pathways altered in hereditary cancer syndromes, which suggests that these pathways are critical to normal cell growth, cell cycle, and proliferation.
SYMBOL | NAME | TUMOR TYPES (GERMLINE MUTATIONS) | CANCER SYNDROME |
|---|---|---|---|
ALK | anaplastic lymphoma kinase (Ki-1) | Neuroblastoma | Familial neuroblastoma |
APC | adenomatous polyposis of the colon gene | Colorectal, pancreatic, desmoid, hepatoblastoma, glioma, other CNS | Adenomatous polyposis coli; Turcot syndrome |
ATM | ataxia telangiectasia mutated | Leukemia, lymphoma, medulloblastoma, glioma | Ataxia-telangiectasia |
BLM | Bloom Syndrome | Leukemia, lymphoma, skin squamous cell, other cancers | Bloom Syndrome |
BMPR1A | bone morphogenetic protein receptor, type IA | Gastrointestinal polyps | Juvenile polyposis |
BRCA1 | familial breast/ovarian cancer gene 1 | Breast, ovarian | Hereditary breast/ovarian cancer |
BRCA2 | familial breast/ovarian cancer gene 2 | Breast, ovarian, pancreatic | Hereditary breast/ovarian cancer |
BRIP1 | BRCA1 interacting protein C-terminal helicase 1 | AML, leukemia, breast | Fanconi anaemia J, breast cancer susceptiblity |
BUB1B | BUB1 budding uninhibited by benzimidazoles 1 homolog beta (yeast) | Rhabdomyosarcoma | Mosaic variegated aneuploidy |
CDH1 | cadherin 1, type 1, E-cadherin (epithelial) (ECAD) | Gastric, lobular cancer | Familial gastric carcinoma |
CDK4 | cyclin-dependent kinase 4 | Melanoma | Familial malignant melanoma |
CDKN2A | cyclin-dependent kinase inhibitor 2A (p16(INK4a)) gene | Melanoma, pancreatic | Familial malignant melanoma |
CDKN2a(p14) | cyclin-dependent kinase inhibitor 2A– p14ARF protein | Melanoma, pancreatic | Familial malignant melanoma |
CHEK2 | CHK2 checkpoint homolog (S. pombe) | Breast | Familial breast cancer |
CYLD | familial cylindromatosis gene | Cylindroma | Familial cylindromatosis |
DDB2 | damage-specific DNA binding protein 2 | Skin basal cell, skin squamous cell, melanoma | Xeroderma pigmentosum (E) |
DICER1 | dicer 1, ribonuclease type III | Pleuropulmonary blastoma | Familial Pleuropulmonary Blastoma |
EGFR | epidermal growth factor receptor (erythroblastic leukemia viral (v-erb-b) oncogene homolog, avian) | NSCLC | Familial lung cancer |
ERCC2, 3, 4, 5 | excision repair cross-complementing rodent repair deficiency, complementation group | Skin basal cell, skin squamous cell, melanoma | Xeroderma pigmentosum (D, B, F, G)) |
EXT1 | multiple exostoses type 1 gene | exostoses, osteosarcoma | exostoses, osteosarcoma |
FANCA, C, D2, E, F, G | Fanconi anemia, complementation group | AML, leukemia | Fanconi anaemia A, C, D2, E, F, G |
FH | fumarate hydratase | leiomyomatosis, renal | Hereditary leiomyomatosis and renal cell cancer |
GPC3 | glypican 3 | Wilms’ tumor | Simpson-Golabi-Behmel syndrome |
HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog | v-Ha-ras Harvey rat sarcoma viral oncogene homolog | Costello syndrome |
HRPT2 | Hyperparathyroidism 2 (parafibromin) | parathyroid adenoma, mulitiple ossifying jaw fibroma | Hyperparathyroidism-jaw tumor syndrome |
KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | GIST, epithelioma | Familial gastrointestinal stromal tumor |
MADH4 | Homolog of Drosophila Mothers Against Decapentaplegic 4 gene | Gastrointestinal polyps | Juvenile polyposis |
MEN1 | multiple endocrine neoplasia type 1 gene | Parathyroid adenoma, pituitary adenoma, pancreatic islet cell, carcinoid | Parathyroid adenoma, pituitary adenoma, pancreatic islet cell, carcinoid |
MLH1 | E. coli MutL homolog gene | Colorectal, endometrial, ovarian, CNS | Hereditary nonpolyposis colorectal cancer, Turcot syndrome |
MPL | myeloproliferative leukemia virus oncogene, thrombopoietin receptor | MPD | Familial essential thrombocythemia |
MSH2 | mutS homolog 2 (E. coli) | colorectal, endometrial, ovarian | Hereditary non-polyposis colorectal cancer |
MSH6 | mutS homolog 6 (E. coli) | colorectal, endometrial, ovarian | Hereditary non-polyposis colorectal cancer |
MUTYH | mutY homolog (E. coli) | Colorectal | Adenomatous polyposis coli |
NBS1 | Nijmegen breakage syndrome 1 (nibrin) | NHL, glioma, medulloblastoma, rhabdomyosarcoma | Nijmegen breakage syndrome |
NF1 | neurofibromatosis type 1 gene | Neurofibroma, glioma | Neurofibromatosis type 1 |
NF2 | neurofibromatosis type 2 gene | Meningioma, acoustic neuroma | Neurofibromatosis type 2 |
PALB2 | partner and localizer of BRCA2 | Wilms tumor, medulloblastoma, AML, breast | Fanconi anaemia N, breast cancer susceptibility |
PHOX2B | paired-like homeobox 2b | Neuroblastoma | Familial neuroblastoma |
PMS1 | PMS1 postmeiotic segregation increased 1 (S. cerevisiae) | Colorectal, endometrial, ovarian | Hereditary non-polyposis colorectal cancer |
PMS2 | PMS2 postmeiotic segregation increased 2 (S. cerevisiae) | Colorectal, endometrial, ovarian, medulloblastoma, glioma | Hereditary nonpolyposis colorectal cancer, Turcot syndrome |
PRKAR1A | protein kinase, cAMP-dependent, regulatory, type I, alpha (tissue specific extinguisher 1) | Myxoma, endocrine, papillary thyroid | Carney complex |
PTCH | Homolog of Drosophila Patched gene | Skin basal cell, medulloblastoma | Nevoid Basal Cell Carcinoma Syndrome |
PTEN | phosphatase and tensin homolog gene | Hamartoma, glioma, prostate, endometrial | Cowden Syndrome, Bannayan-Riley-Ruvalcaba syndrome |
RB1 | retinoblastoma gene | Retinoblastoma, sarcoma, breast, small cell lung | Familial retinoblastoma |
RECQL4 | RecQ protein-like 4 | Osteosarcoma, skin basal and squamous cell | Rothmund-Thompson Syndrome |
RET | ret proto-oncogene | Medullary thyroid, papillary thyroid, pheochromocytoma | Multiple endocrine neoplasia 2A/2B |
SBDS | Shwachman-Bodian-Diamond syndrome protein | AML, MDS | Schwachman-Diamond syndrome |
SDH5 | chromosome 11 open reading frame 79 | Paraganglioma | Familial paraganglioma |
SHD, B, D | succinate dehydrogenase complex | Paraganglioma, pheochromocytoma | Familial paraganglioma |
SMARCB1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 | Malignant rhabdoid | Rhabdoid predisposition syndrome |
STK11 | serine/threonine kinase 11 gene (LKB1) | Jejunal hamartoma, ovarian, testicular, pancreatic | Peutz-Jeghers syndrome |
SUFU | suppressor of fused homolog (Drosophila) | Medulloblastoma | Medulloblastoma predisposition |
TCF1 | transcription factor 1, hepatic (HNF1) | Hepatic adenoma, hepatocellular carcinoma | Familial Hepatic Adenoma |
TP53 | tumor protein p53 | Breast, sarcoma, adrenocortical carcinoma, glioma, multiple other tumor types | Li-Fraumeni syndrome |
TSC1 | tuberous sclerosis 1 gene | Hamartoma, renal cell | Tuberous sclerosis 1 |
TSC2 | tuberous sclerosis 2 gene | Hamartoma, renal cell | Tuberous sclerosis 2 |
TSHR | thyroid stimulating hormone receptor | Thyroid adenoma | |
VHL | von Hippel-Lindau syndrome gene | Renal, hemangioma, pheochromocytoma | von Hippel-Lindau syndrome |
WRN | Werner syndrome (RECQL2) | Osteosarcoma, meningioma, others | Werner Syndrome |
WT1 | Wilms’ tumor 1 gene | Wilms’ | Denys-Drash syndrome, Frasier syndrome, Familial Wilms tumor |
XPA, C | xeroderma pigmentosum, complementation group | Skin basal cell, skin squamous cell, melanoma | Xeroderma pigmentosum (A C) |
The following factors may suggest the presence of a hereditary cancer49:
Tumor development at a much younger age than usual
Presence of bilateral disease
Presence of multiple primary malignancies
Presentation of a cancer in the less affected sex (e.g., male breast cancer)
Clustering of the same cancer type in relatives
Occurrence of cancer in association with other conditions such as mental retardation or pathognomonic skin lesions
It is crucial that all surgeons caring for cancer patients be aware of hereditary cancer syndromes, because a patient’s genetic background has significant implications for patient counseling, planning of surgical therapy, and cancer screening and prevention. Some of the more commonly encountered hereditary cancer syndromes are discussed here.
The retinoblastoma gene rb1 was the first tumor suppressor to be cloned. The rb1 gene product, the Rb protein, is a regulator of transcription that controls the cell cycle, differentiation, and apoptosis in normal development.50 Retinoblastoma has long been known to occur in hereditary and nonhereditary forms. Interestingly, although most children with an affected parent develop bilateral retinoblastoma, some develop unilateral retinoblastoma. Furthermore, some children with an affected parent are not affected themselves but then have an affected child, which indicates that they are rb1 mutation carriers. These findings led to the theory that a single mutation is not sufficient for tumorigenesis. Alfred Knudson hypothesized that hereditary retinoblastoma involves two mutations, of which one is germline and one somatic, whereas nonhereditary retinoblastoma is due to two somatic mutations (Fig. 10-12).51 Thus, both hereditary and nonhereditary forms of retinoblastoma involve the same number of mutations, a hypothesis known as Knudson’s “two-hit” hypothesis. A “hit” may be a point mutation, a chromosomal deletion referred to as allelic loss, or a loss of heterozygosity, or silencing of an existing gene.
Figure 10-12.
Tumor heterogeneity. A. Patients with tumors with similar histologies may differ in genetic mutation status and other molecular features B. Cells within the primary tumor can acquire or lose genomic alterations in metastatic sites. C. Intratumoral spatial heterogeneity: common initiating genomic events usually exist in all tumor cells but additional spatially separated heterogeneous somatic mutations or copy number changes may accumulate. (Adapted with permission from Meric-Bernstam and Mills)44
Li-Fraumeni syndrome (LFS) was first defined on the basis of observed clustering of malignancies, including early-onset breast cancer, soft tissue sarcomas, brain tumors, adrenocortical tumors, and leukemia.52 Criteria for classic LFS in an individual (the proband) include: (a) a bone or soft tissue sarcoma when younger than 45 years, (b) a first-degree relative with cancer before age 45 years, and (c) another first- or second-degree relative with either a sarcoma diagnosed at any age or any cancer diagnosed before age 45 years.53 Approximately 70% of LFS families have been shown to have germline mutations in the tumor-suppressor gene p53.54 Breast carcinoma, soft tissue sarcoma, osteosarcoma, brain tumors, adrenocortical carcinoma, Wilms’ tumor, and phyllodes tumor of the breast are strongly associated; pancreatic cancer is moderately associated; and leukemia and neuroblastoma are weakly associated with germline p53 mutations.55 Mutations of p53 have not been detected in approximately 30% of LFS families, and it is hypothesized that genetic alterations in other proteins interacting with p53 function may play a role in these families.
Of the known genes in human cancer, p53 is the most commonly mutated. The p53 protein regulates cell-cycle progression as well as apoptotic cell death as part of stress response pathways after exposure to ionizing or ultraviolet (UV) irradiation, chemotherapy, acidosis, growth factor deprivation, or hypoxia. When cells are exposed to stressors, p53 acts as a transcription factor for genes that induce cell-cycle arrest or apoptosis. A majority of p53 mutations are found within a central DNA recognition motif and disrupt DNA binding by p53. Families with germline missense mutations in the DNA-binding domain show a more highly penetrant phenotype than families with other p53 mutations.56 Furthermore, proband cancers are linked with significantly younger age at diagnosis in patients with missense mutations in the DNA-binding domain.56
It is estimated that 5% to 10% of breast cancers are hereditary. Of women with early-onset breast cancer (aged 40 years or younger), nearly 10% have a germline mutation in one of the breast cancer genes BRCA1 or BRCA2.57 Mutation carriers are more prevalent among women who have a first- or second-degree relative with premenopausal breast cancer or ovarian cancer at any age. The likelihood of a BRCA mutation is higher in patients who belong to a population in which founder mutations may be prevalent, such as in the Ashkenazi Jewish population. For a female BRCA1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


