Neuroendocrine tumors (NETs) in childhood and adolescents are uncommon. According to the Surveillance, Epidemiology, and End Results database, every type of NET that occurs in adults also occurs in childhood, although infrequently. In these data, the most common NETs in pediatric and adolescents groups represented neoplasms of neural crest origin such as neuroblastomas. NETs in this age group may be associated with certain hereditary syndromes (
Chapter 1;
Table 1.1). Several of these neuroendocrine neoplasms such as thyroid medullary carcinomas, pheochromocytomas, pituitary adenomas, and gastrointestinal and pancreatic NETs have already been described in previous chapters and are not repeated. This chapter describes frequently encountered NETs in childhood and adolescents, which represent neuroblastic tumors such as neuroblastoma, ganglioneu-roblastoma and ganglioneuroma, and Ewing sarcoma/peripheral neuroectodermal tumors. Olfactory neuroblastoma can occur rarely in the pediatric age group and is discussed in
Chapter 17.
The application of diagnostic procedures such as fine needle aspiration (FNA) biopsy is generally underutilized in large Pediatric Centers. Overall, the exposure to cytopathology of pediatric tumors for most pathologists/cytopathologists is rather modest. The author has made serious efforts to cover the subject in fair detail.
NEUROBLASTOMA
Neuroblastomas, ganglioneuroblastomas, and ganglioneuromas arise from primitive cells of neural crest origin and represent maturational stages of a common neoplasm. The first two being malignant are considered together, while ganglioneuroma, the differentiated neuroblastic neoplasm, is described separately.
Neuroblastoma/ganglioneuroblastoma is one of the common solid tumors of infancy and childhood and the fourth most common malignancy in this age group, after leukemia, brain tumors, and malignant lymphomas. They represent 10% to 20% of all malignant pediatric tumors. One-fourth of the cases are congenital. The annual incidence in the United States is reported to be 9.2 per million children. The incidence is higher in Japan, reported as 19 per million children as a result of prenatal screening.
CLINICAL FEATURES
Neuroblastomas occur in various paraspinal locations and in parenchymal organs, the most frequent sites being adrenal medulla, extra-adrenal retroperitoneal areas, mediastinum, cervical ganglia, and head and neck region. The majority is diagnosed in children <5 years of age. There is no gender predilection. The presenting symptoms depend on location of the tumor and presence or absence of associated syndromes. Children with neuroblastomas often appear systemically sick, wasted, and with nonspecific symptoms, or the symptoms may be related to the metastatic disease. Patients may have blue-red cutaneous metastatic lesions, present with Horner syndrome, or have opsoclonus-myoclonus syndrome (rapid alternating eye movements followed by myoclonic movements of extremities). Paraneoplastic syndromes such as vasoactive intestinal polypeptide syndrome with chronic watery diarrhea, abdominal distension, and hypertension are not rare. A palpable abdominal mass is noted in a significant number of patients.
Laboratory studies include high urinary levels of catecholamines and their metabolites that are characteristic findings. Neuroblastomas can metastasize widely, to liver and bones, including orbit, maxillofacial bones, and paranasal sinuses.
The biologic behavior of neuroblastomas varies from excellent to fatal outcome. It is dependent on the age at detection, histologic grade, extent of the metastatic spread, and the stage of the disease. Two-year survival rates vary from 100% to 18%.
RADIOLOGIC FINDINGS
A mass lesion along the sympathetic trunk or in the adrenal medulla is the most common finding on CT or MRI, with the adrenal gland being the most common site. The mass often shows calcifications. Metastatic lesions in bones are osteolytic and have a predilection for the skull, femur, and humerus.
GROSS AND MICROSCOPIC FEATURES
Grossly, neuroblastomas vary in size and consistency. They are often well-circumscribed with a lobulated contour. They may be reddish purple to grayish homogenous with microcalcifications.
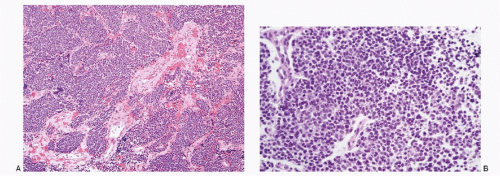
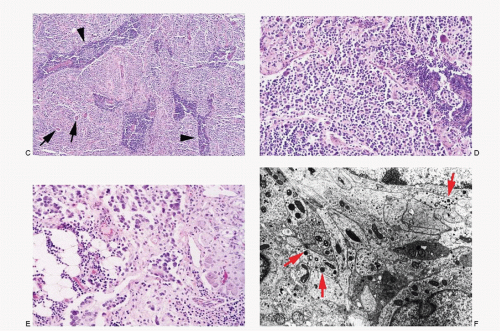
Histomorphology of neuroblastomas presents a wide spectrum dependent on the maturation of the neuroblastic cells (
Fig. 16.1A-E). The classic pattern consists of uniform, small round cells, with large hyperchromatic nuclei, arranged in nests, separated by fibrovascular septae. The cells may demonstrate rosettes (Homer-Wright type). Mitoses are not frequent. Ganglioneuroblastomas consist of variable proportion of ganglion cells and neuroblastic cells (see
Fig. 16.1C-E). Two types of ganglioneuroblastomas are described—nodular and intermixed. The nodular type shows grossly visible nodules of neuroblastic tissue separated by schwannomatous stroma containing spindle-shaped Schwann cells. The intermixed type shows diffuse population of neuroblastic cells and ganglionic cells in varying stages of maturation.
The ganglion cells are identified by their large size, round-to-polygonal shape, and abundant granular cytoplasm. They may be mononucleated, binucleated, or multinucleated with coarsely granular chromatin and a prominent macronucleolus. Neuroblastomas often show fibrillar or bubbly intercellular matrix representing neural processes, referred to as neuropil.
IMMUNOPROFILE
The neuroblastoma cells react positively to all neuroendocrine markers—neuron-specific enolase, chromogranin, and synaptophysin. They also react positively to protein gene product PCP 9.5, CD56, CD57 (Leu 7), and NB-84. Neuroblastomas do not react to GFAP, myelin basic protein, and CD99.
ULTRASTRUCTURE
The neuroblastoma cells present neural differentiation in the form of dendritic processes that contain parallel arrays of microtubules and dense core secretory granules (
Fig. 16.1F). The dense core granules are found in small aggregates in the elongated cell processes as well as in the cell body. The neural processes form interweaving matted medusa-like aggregates, particularly in the central portions of the rosettes.
MOLECULAR/CYTOGENETIC FINDINGS
Neuroblastomas show amplification of MYCN and consistent loss of heterozygosity of 1p and 11q.
CYTOPATHOLOGIC FEATURES
The specimens for cytologic diagnosis mostly represent FNA biopsies at either the primary or metastatic sites. Crush preparations of the tissues submitted for intraoperative consultation and body cavity fluids have also been useful in cytologic diagnosis.
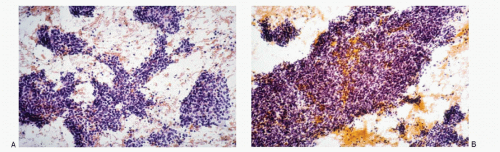
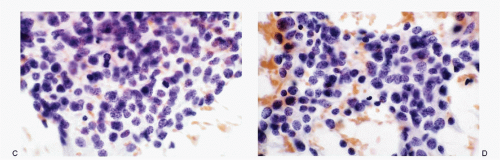
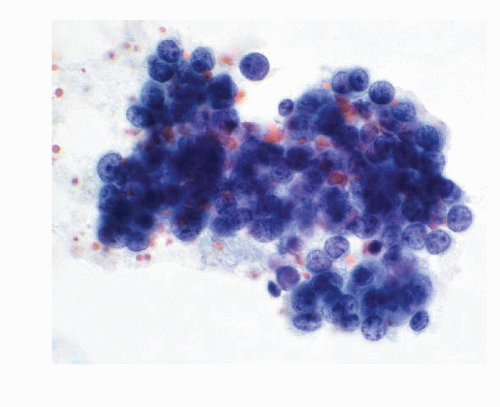
The cytologic features of neuroblastomas (
Table 16.1) depend on the differentiation of the tumor, ranging from undifferentiated cells to the cells with various stages of maturation toward ganglion cells. The aspirates are usually highly cellular consisting of a large population of small round cells with poorly defined cell borders and scant cytoplasm with high N/C ratios (
Figs. 16.2,
16.3,
16.4,
16.5,
16.6 and
16.7). The nuclei are round to oval with deep-staining chromatin, often presenting the typical salt-pepper pattern. Nucleoli are inconspicuous. Nuclear molding may be present. The cells form syncytial tissue fragments with or without rosette formations (Homer-Wright type). The rosettes consist of central matrix or neuropil surrounded by neuroblastoma cells. The latter may possess unipolar cytoplasmic process or tailing. The presence of ganglion cells indicates a maturation process. The background may contain neurofibrillar matrix or neuropil.
DIFFERENTIAL DIAGNOSES
The differential diagnoses of neuroblastoma include small round-cell tumors of childhood (
Table 16.2;
Figs 16.8,
16.9,
16.10,
16.11,
16.12,
16.13,
16.14,
16.15 and
16.16). The presence of rosettes, neuropil, and ganglion cells offers support to the diagnosis of neuroblastoma. The difficulties arise when these features are not present and the cells are totally undifferentiated.
Neuroblastic tumors frequently contain lymphoid cell aggregates that may result in diagnostic problems in differentiating primitive cells. Immunostains for CD45 is helpful in identifying the lymphoid cells.