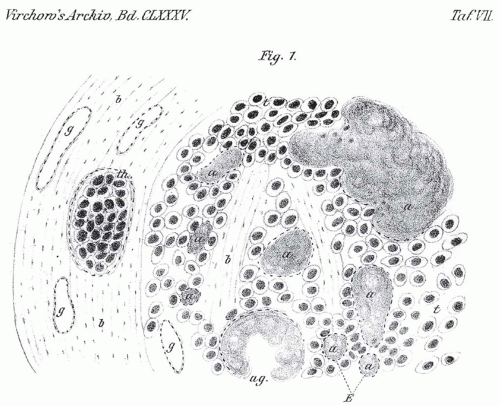
 FIGURE 14.1. Drawing from Jaquet’s 1906 publication regarding an apparent case of medullary thyroid carcinoma. The drawing depicts a metastatic lesion in the mediastinum with a uniform population of tumor cells and deposits of amyloid. The letter key to his diagram is as follows: a, amyloid; t, tumor cells; g, blood vessels; th, tumor thrombus in blood vessel; b, connective tissue; ag, blood vessel transformed by amyloid; E, endothelial membrane. (From Jacquet J. Ein Fall von metastasierenden Amyloidtumoren (Lymphosarkom). Virchows Arch Pathol Anat. 1906;185:251-268.) |
category, but this error is offset by the reduced likelihood of overlooking a pheochromocytoma.
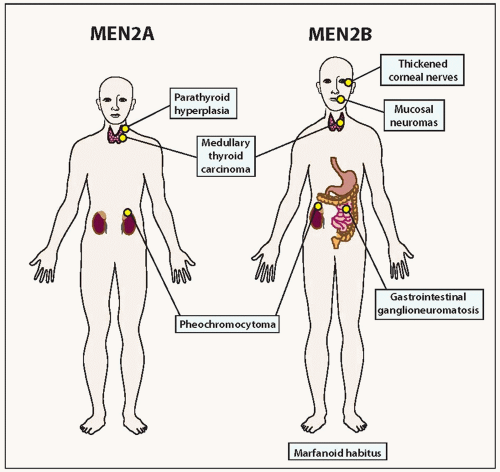 FIGURE 14.2. Characteristic abnormalities associated with MEN2A and MEN2B. |
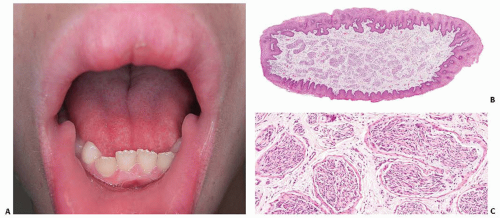 FIGURE 14.3. Oral mucosal neuromas in the lips and tongue of a child with MEN2B. A: Nodules in both oral commissures, the upper lip and the tip of the tongue. B and C: Low-power (B) and medium-power (C) photomicrographs of a biopsy showing numerous, irregular nerve bundles. |
activation of the monomeric form of RET.36,54 A conformational change in the binding pocket of the tyrosine kinase domain leads to an altered substrate specificity. A small portion of FMTC cases have mutations involving the intracellular tyrosine kinase domain. The activation mechanisms associated with these mutations are not well defined at this time.
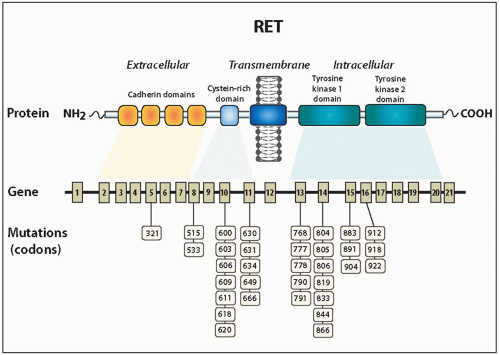 FIGURE 14.4. Schematic diagram of RET gene and associated RET protein. Codons reported to be sites of RET mutations resulting in hereditary medullary thyroid carcinoma are listed below their associated exon. The association between exons and the regions of the RET protein they encode is also shown. |
variation in regard to clinical manifestations of disease. Several potential oncoproteins and tumor suppressors are present in the signaling pathways utilized by the RET receptor, and mutations in genes encoding these proteins may play significant roles in the development of medullary carcinoma. Major signaling proteins activated downstream of RET are RAS and phosphatidylinositol 3-kinase. As discussed above, RAS mutations have been found in a significant number of sporadic medullary carcinomas. Loss
of function of negative regulators of RET signaling such as the tyrosine phosphatases LAR, PTPRJ, or SHP-1 is another potential mechanism of tumorigenesis. Murine studies suggest that mutations of the tumor suppressor genes RB1 and TP53 may play a role in medullary carcinoma.
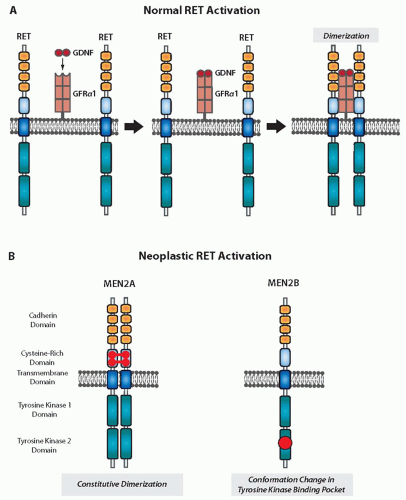 FIGURE 14.5. A: Drawing showing the normal binding of GDNF to GDNF family receptor α1 and subsequent binding of the complex with RET receptor and activation through dimerization. B: Drawings showing constitutive dimerization of RET in MEN2A and FTMC and monomeric activation in MEN2B due to change in tyrosine kinase binding pocket. |
Table 14.1 Sites of RET Germline Mutations and Associated Forms of Hereditary Medullary Thyroid Carcinoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
C cells are usually credited to E. Cresswell Baber79,80 who in 1876 and 1878 described them within the dog thyroid. He noted their parafollicular location and different appearance from follicular cells, calling them parenchymatous cells. In 1894, Hürthle81 provided additional description of what he thought were identical cells and applied the term protoplasm-rich cells (protoplasmareichen Zellen) (Fig. 14.6). Although it appears that Hürthle described C cells, his name is often linked to metaplastic oncocytic follicular cells instead.
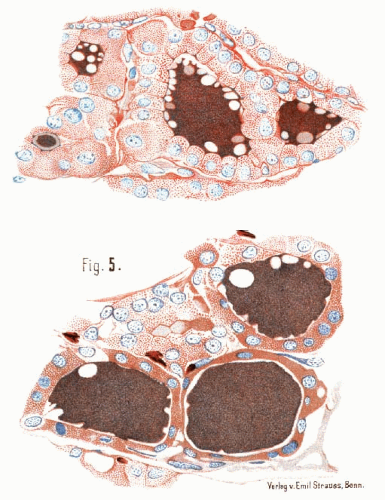 FIGURE 14.6. Illustrations from Hürthle’s 1894 article describing protoplasm-rich cells in the dog thyroid gland, which are now thought to represent C cells. The upper illustration shows the cells wedged between follicular cells, whereas the lower illustration shows them in a parafollicular location. (From Hürthle K. Beiträge zur Kenntniss des Secretionsvorgangs in der Schilddrüse. Arch Gesammte Physiol. 1894;56:1-44.) |
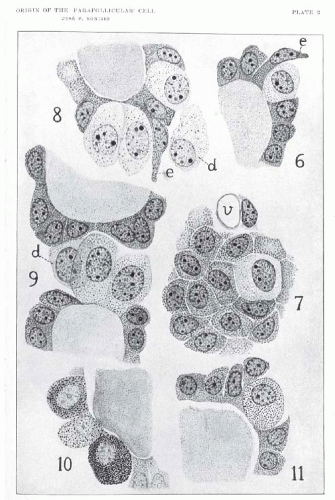 FIGURE 14.7. Illustration from Nonidez’s 1932 article demonstrating argyrophilic granules within the cytoplasm of parafollicular cells of the dog thyroid gland. Parafollicular cells are designated “d” and elongated follicular cells “e.” (From Nonidez JF. The origin of the “parafollicular” cell, a second epithelial component of the thyroid gland of the dog. Am J Anat. 1932;49:479-505.) |
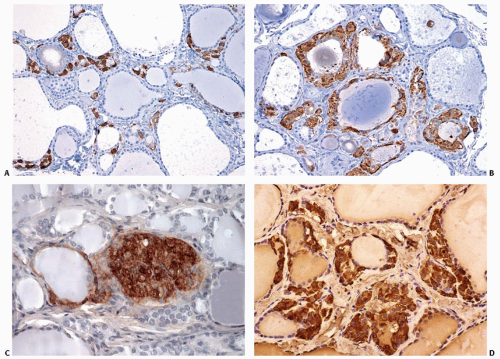 FIGURE 14.8. Growth patterns of C-cell hyperplasia. A: Focal hyperplasia with segmental proliferation. B: Focal and diffuse hyperplasia (encirclement of follicles). C: Nodular hyperplasia with obliteration of follicular lumen. D: Diffuse and nodular hyperplasia with central area suspicious for invasion beyond follicular basement membrane (early medullary carcinoma). |
genetic testing is advisable because of potential identification of a new hereditary kindred.
Table 14.2 C-Cell Hyperplasia—Definitions of Subtypes and Growth Patterns | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
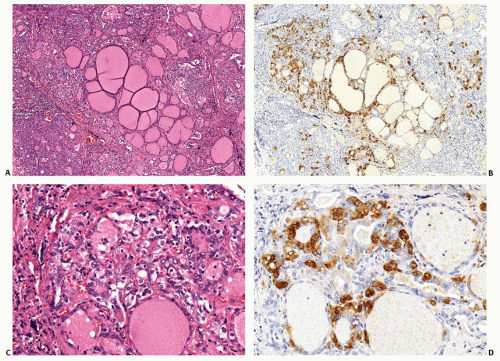 FIGURE 14.9. C-cell hyperplasia associated with chronic lymphocytic (Hashimoto) thyroiditis. A: Low-power view of H&E-stained section showing features of chronic lymphocytic thyroiditis. B: Calcitonin-stained section of same area as (A) showing numerous C cells. C: High-power view of H&E-stained section showing C cells with clear cytoplasm. D: High-power view of calcitonin-stained section showing encirclement of follicles by C cells. |
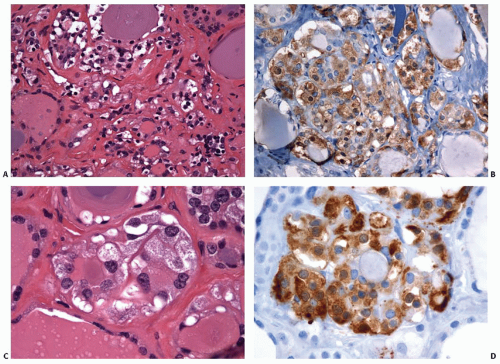 FIGURE 14.10. “Neoplastic” C-cell hyperplasia. Large C cells with clear or lightly stained cytoplasm detectable in H&E-stained sections (A and C). Calcitonin-immunostained sections of same areas (B and D). |
combined with 10× eyepiece) is the most common criterion, even this has its variations as requirements range from this number of cells in just one field, to at least one field in each lobe, or to at least three fields in either or both lobes. Several other criteria have been used in the past.105
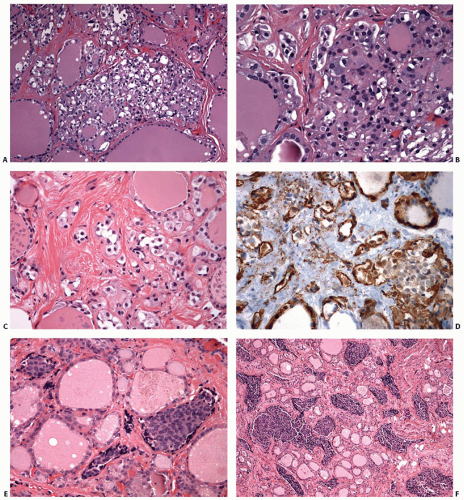 FIGURE 14.11. A-D: Two cases of early medullary microcarcinoma arising in an area of “neoplastic” C-cell hyperplasia. A fibrotic response to cells extending beyond the follicles is seen. E and F: Multifocal intrathyroidal spread of medullary carcinoma. Small isolated foci bear resemblance to nodular C-cell hyperplasia (A-C, E, and F, H&E; D, calcitonin immunostain). |
problematic when a high percentage of the “normal” population meets the criterion.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


