Mechanisms of Biofilm Formation in the Staphylococci
Paul D. Fey
As documented in other chapters of this text, both Staphylococcus aureus and S. epidermidis are a cause of significant infections in both the community and the hospital environment. Most likely due to their ability to colonize mucous membranes and skin of humans, data from the National Nosocomial Infection Surveillance (NNIS) system demonstrate that S. aureus and S. epidermidis are the most common cause of healthcare-associated infections including catheter-related infections (1). One particularly onerous aspect of staphylococcal pathogenesis is the ability of most staphylococcal species to form biofilm on foreign medical devices (2). Despite the documented function of biofilm in the context of staphylococcal disease, our understanding of molecular mechanisms fundamental to biofilm formation is incomplete. It is clear that bacteria within a biofilm have a unique and heterogeneous metabolism compared to planktonic cells (3). Consequently, bacterial biofilms are inherently resistant to antibiotics and the innate immune system leaving clinicians little choice but to remove the infected device. Staphylococcal biofilm is defined as a heterogeneous mixture of cells, usually bound to a foreign body, encased in an extracellular matrix consisting of protein, extracellular DNA (eDNA), and polysaccharide (2,4). Recent data from a variety of investigators have clearly shown that extracellular matrix differs between strains; in fact, the composition of the extracellular matrix within a particular isolates biofilm may change depending upon the extracellular niche (5, 6 and 7). In an attempt to organize the data, this chapter will focus on the four identified stages of S. aureus and S. epidermidis biofilm synthesis (a) initial attachment, (b) accumulation, (c) maturation processes, and (d) dispersion. However, in some cases, particular factors clearly function in multiple stages of biofilm formation (i.e., initial attachment and accumulation). Particular biofilm components may have been studied in the context of either S. aureus or S. epidermidis biofilm, and this will be noted.
STAPHYLOCOCCAL ATTACHMENT TO A FOREIGN BODY
Both S. aureus and S. epidermidis have multiple factors that function to mediate adherence to foreign biomaterials. After insertion into a host, foreign medical devices are rapidly coated by serum proteins including fibrinogen, fibronectin, collagen, and vitronectin. Staphylococci have the capability to bind these serum proteins, and thus the biomaterial, through the interaction with MSCRAMMs (Microbial Surface Components Recognizing Adhesive Matrix Molecules) (8, 9 and 10). Although the number of MSCRAMMs encoded by a particular strain differs, S. aureus (˜20) typically encodes more MSCRAMM-like proteins than does S. epidermidis (˜12) (8,11). MSCRAMMs have a common structure including a surface-exposed-binding domain, a cell wall-spanning domain, and a peptidoglycan-binding domain that is typically covalently attached through an LPXTG motif by sortase A (12,13). S. aureus MSCRAMM examples include the fibronectin/fibrinogen/elastin-binding proteins FnBpA and FnBpB (14), two unique fibrinogen-binding proteins ClfA and ClfB (15,16), and the collagen-binding protein Cna (17). Other staphylococcal MSCRAMMs found in both S. aureus and S. epidermidis include a family of proteins containing extensive serine-aspartate (SD) repeats (8). One extensively studied MSCRAMM in S. epidermidis and S. aureus is SdrG, a molecule that binds fibrinogen through a “dock, lock, and latch” mechanism (12). Evidence also suggests that (GehD), a cell wall-associated protein that does not encode an LPXTG motif, is a bifunctional enzyme that also binds collagen in addition to its activity as a lipase (18).
In addition to binding specifically to serum proteins, staphylococci, especially S. epidermidis, have the unique ability to bind plastic polymers (e.g., catheters). Heilmann and colleagues found this binding property is linked to the autolysin AtlE in S. epidermidis (19). It was subsequently shown that atlE mutants of S. epidermidis were less virulent in a rat vascular catheter model (20). Further work demonstrated that both AtlE bind vitronectin whereas a separate autolysin, Aae, binds fibrinogen, fibronectin, and vitronectin in a dose-dependent manner (19,21). Thus, autolysins of staphylococci have the ability to bind both polymers (through an unknown mechanism) and serum proteins. Somewhat complicating the interpretation of the autolysin mutant phenotype, however, is the recent finding of the importance of bacterial extracellular DNA (eDNA) in the establishment of a staphylococcal biofilm (22,23). eDNA provides structure that is important for both initial adherence and biofilm accumulation; thus, bacteria that are unable to lyse effectively (atlE or aae mutants) would be defective in eDNA release and thus be less adherent.
Thus, autolysins may have multiple roles in biofilm formation including the release of eDNA, initial adherence to polymers, and specific binding to serum proteins.
Thus, autolysins may have multiple roles in biofilm formation including the release of eDNA, initial adherence to polymers, and specific binding to serum proteins.
STAPHYLOCOCCAL BIOFILM ACCUMULATION
Defining the multiple mechanisms of staphylococcal biofilm accumulation and its clinical relevance has been an extremely exciting avenue of recent investigation. Recent data have suggested that S. epidermidis biofilm accumulation is mediated mostly by polysaccharide intercellular adhesin (PIA) and is sensitive to polysaccharide-dispersing enzymes such as dispersin B but resistant to proteases, whereas the converse is true for S. aureus biofilms (24). However, there is increasing evidence demonstrating that clinical isolates of S. epidermidis produce proteinaceous biofilms (i.e., accumulation associated protein [Aap]) (5). Furthermore, antibodies against PIA (or PNAG; poly-N-acetyl glucosamine) are protective in animal models of S. aureus infection suggesting expression of PIA/PNAG in vivo (25). Therefore, the available data suggest that multiple proteins and PIA/PNAG may function to mediate biofilm accumulation within staphylococci, and their expression may depend on the nutritional state of the bacterium and/or niche. It is unknown whether the expression of these unique accumulation molecules exhibits any clinical significance in regards to clinical management of the biofilm-mediated infection or whether the metabolism differs in a PIA/PNAG-dependent biofilm in comparison to a proteinaceous-sensitive biofilm. Understanding these and other questions is fundamental to the rational design of new antibiotics and/or biofilm inhibitors. This section will summarize the molecules known to mediate biofilm accumulation; transcriptional regulation of these determinants will be discussed if known.
Polysaccharide Intercellular Adhesin/Poly-N-Acetylglucosamine
Staphylococcal biofilm accumulation has been most well studied in the context of polysaccharide intercellular adhesin (PIA) or poly-N-acetylglucosamine (PNAG) synthesis. PIA is a β-1,6 linked poly-N-acetylated glucosamine and its synthesis is directed by enzymes encoded by the icaADBC operon (26,27). An identical molecule has been identified from S. aureus and is termed PNAG (28,29). All four open reading frames (ORFs; icaA, icaD, icaB, and icaC) are required for PIA/PNAG synthesis (30,31). Utilizing UDP-N-acetylglucosamine as a substrate, IcaAD acts as an N-acetylglucosaminyl transferase; IcaC, a membrane protein, most likely functions to transfer the growing polysaccharide outside of the bacterium to the cell wall. IcaB is a secreted deacetylase that functions to deacetylate PIA/PNAG; 15% to 43% of glucosamine residues are deacetylated (29,32). Vuong and colleagues found that a 1457 icaB mutant was unable to form functional biofilm and exhibited reduced virulence in a foreign body infection model demonstrating the importance of deacetylation in PIA/PNAG-mediated biofilm accumulation (33). Utilizing icaADBC mutants, several studies have demonstrated the importance of PIA in virulence of S. epidermidis using relevant animal models of infections; these studies found that PIA-deficient strains of S. epidermidis 1457 and O-47 have reduced virulence in comparison to isogenic PIA positive strains (20,34,35,36). Furthermore, PIA/PNAG inhibits neutrophil-dependent killing and mediates biocide resistance (37,38). However, it is important to note and as will be detailed below, multiple clinical strains of S. epidermidis do not encode the icaADBC operon (39, 40 and 41,42,43) and, even when encoded, icaADBC can be highly repressed (unpublished observations from the author). Similarly, most studies have found icaADBC encoded in almost all S. aureus isolates (30,39,44), but the operon is highly repressed and is dispensable for biofilm formation in vitro (39,45, 46, 47 and 48). However, icaADBC expression is upregulated in S. aureus during infection demonstrating that icaADBC expression is strain dependent and contains multiple layers of transcriptional and/or translational regulation (47,49). Transcriptional regulation of icaADBC is very complex and published reports have documented 14 unique direct or indirect regulatory elements (Fig. 31-1).
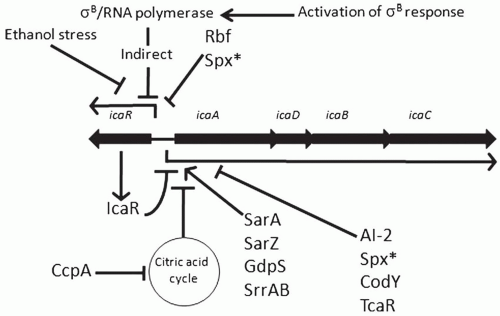 FIGURE 31-1 icaADBC regulatory circuit. Regulatory proteins known to function as activators of either icaADBC or icaR transcription are indicated with an arrow. Those that are known to repress transcription of icaADBC or icaR are indicated by a blunt arrow. (*) Spx is known to repress icaR transcription in S. aureus but represses icaADBC in S. epidermidis. |
σB—The first regulatory element identified as a member of the icaADBC regulon was σB, the alternative sigma factor in staphylococci (50). Insertion of Tn917 into the S. epidermidis 1457 rsbU gene, a positive regulator of σB expression, led to significantly decreased biofilm and PIA synthesis. Further work demonstrated that σB functions in an indirect manner to repress expression of icaR, a transcriptional repressor of icaADBC (51); inactivation of
σB led to increased expression of IcaR and thus, decreased icaADBC transcription (52,53). Ethanol pressure is also known to repress icaR transcription, however, this regulatory pathway is σB independent (54).
σB led to increased expression of IcaR and thus, decreased icaADBC transcription (52,53). Ethanol pressure is also known to repress icaR transcription, however, this regulatory pathway is σB independent (54).
IcaR/TcaR—icaR, a member of the TetR family of transcriptional regulators, is divergently transcribed from icaADBC (Fig. 31-1) and negatively regulates icaADBC transcription (51). IcaR functions as a dimer and binds cooperatively to a 28-bp region upstream of icaADBC (55). Cerca and colleagues recently found that in S. aureus, in contrast to S. epidermidis, icaR transcription is SarA and σB dependent, and IcaR is not required for activation of its own transcription suggesting that S. aureus and S. epidermidis regulate icaR transcription differently (56). Utilizing the ica promoter region as a target in pull-down assays, Jefferson and colleagues identified a second regulator, TcaR, which has recently been shown to bind specifically to three separate regions in the icaADBC promoter region (57,58). Interestingly, similar to IcaR, antibiotics bind to TcaR and inhibit their binding to the promoter region thus activating icaADBC transcription and PIA/PNAG production (55,57).
Rbf—Rbf (Regulator of Biofilm Formation) was first identified as a locus that regulated biofilm formation in response to glucose and sodium chloride in S. aureus (59). Rbf has homology to the AraC/XylS family of transcriptional regulators and functions in an indirect manner to regulate icaADBC transcription by repressing icaR transcription (60). As predicted, inactivation of rbf led to decreased virulence of S. aureus in a mouse model of foreign body infection due to decreased PIA/PNAG synthesis, although the effect was strain dependent (61).
SarA/SarZ—The Sar family of DNA-binding proteins is a group of transcriptional regulators that function to activate or repress a large number of genes within the staphylococci, many of which are virulence genes including biofilm formation (62). SarA has consistently been noted as a factor absolutely required for biofilm formation in both PIA/PNAG-dependent and PIA/PNAG-independent biofilms in S. aureus and S. epidermidis (52,63, 64, 65, 66 and 67). SarA is a positive regulator impacting icaADBC transcription in an icaR-independent manner (65,66), although its function in regulating biofilm formation in PIA/PNAG biofilms is not clear but may involve protease production (64). Furthermore, SarZ also acts as a positive activator of icaADBC transcription in S. epidermidis apparently independent of SarA (68), although others have observed a relationship between SarA and SarZ expression and their interaction on biofilm formation in S. aureus (69).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


