|
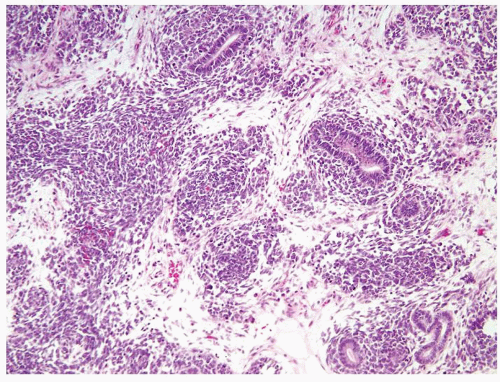 Figure 2.1.1 Wilms tumor displaying triphasic histology (blastemal, stromal, and epithelial). |
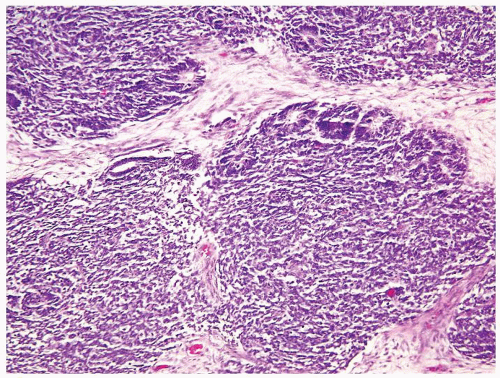 Figure 2.1.2 Wilms tumor with mostly blastemic and focal tubular differentiation at the periphery of the blastema. |
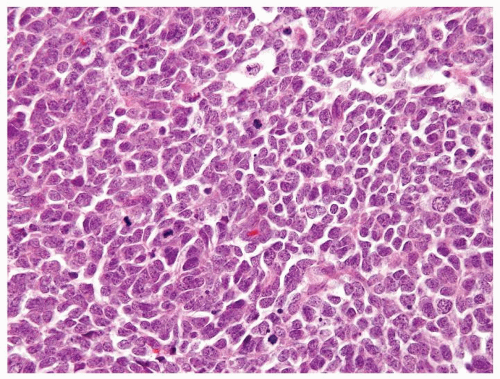 Figure 2.1.3 Wilms tumor with a high nuclear-to-cytoplasmic ratio, coarse chromatin, and numerous mitotic figures. |
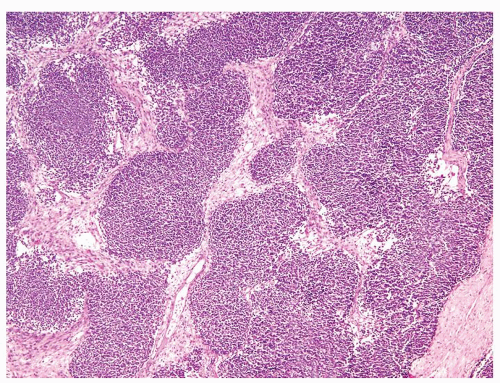 Figure 2.1.4 Wilms tumor with serpentine growth pattern. |
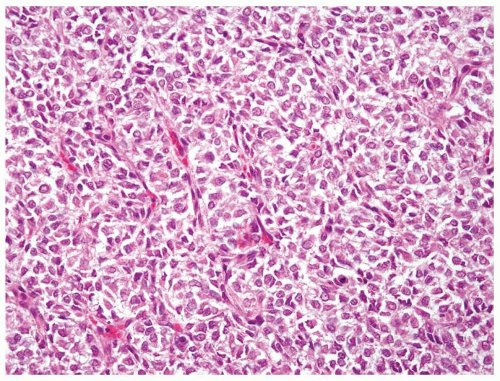 Figure 2.1.5 Classic clear cell sarcoma with nests separated by evenly dispersed small vascular channels. |
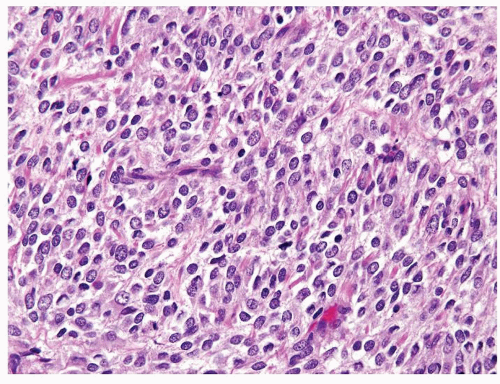 Figure 2.1.6 Clear cell sarcoma with nests separated by small thinwalled vessels. |
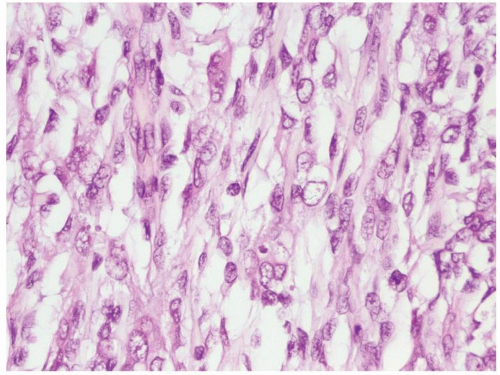 Figure 2.1.7 Clear cell sarcoma with open nuclei and uncommon mitotic figures. |
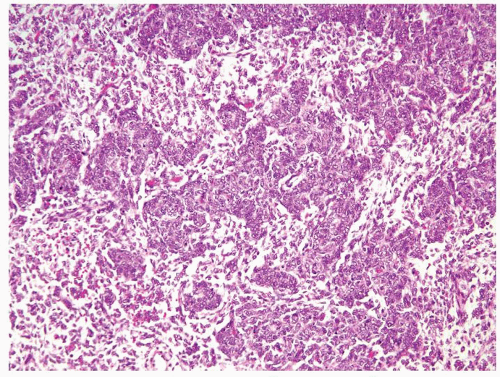 Figure 2.1.8 Clear cell sarcoma with condensed cords of cells with an epithelioid appearance that can mimic Wilms tumor. |
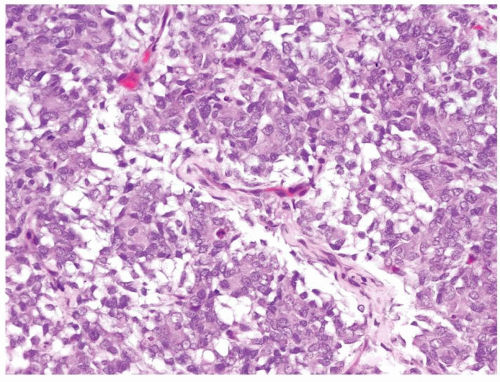 Figure 2.1.9 Clear cell sarcoma with condensed tumor cells in myxoid stroma separated by fine vascular network. |
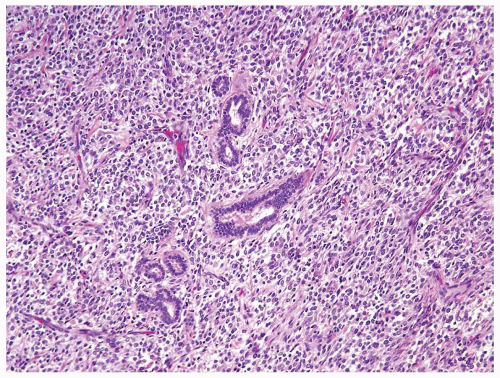 Figure 2.1.10 Clear cell sarcoma with entrapped tubules. |
|
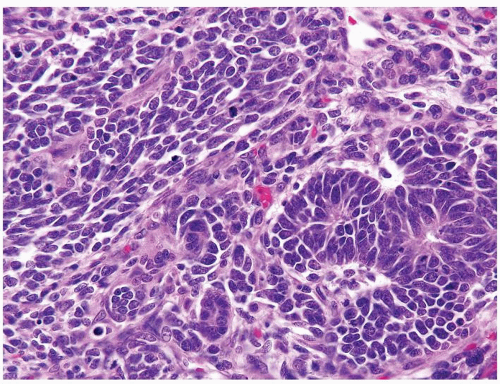 Figure 2.2.1 Wilms tumor with tubules and blastema. The blastema has a high mitotic rate. The tubules are lined by overlapping nuclei with a high N/C ratio. |
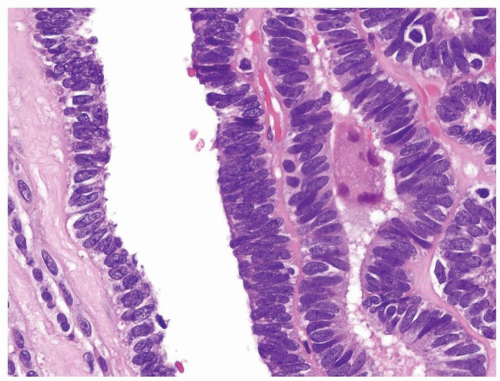 Figure 2.2.2 Wilms tumor with tubules having atypical elongated nuclei with an overlapping and high N/C ratio. |
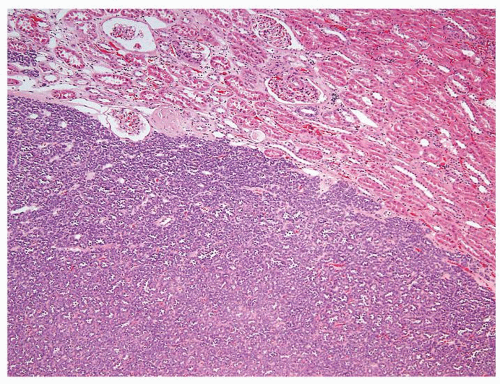 Figure 2.2.3 Metanephric adenoma showing well-circumscribed border and “blue,” low-power appearance. |
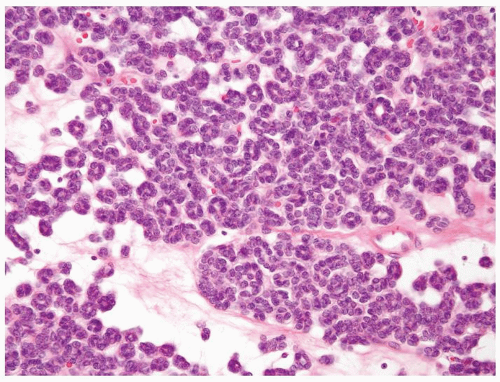 Figure 2.2.4 Metanephric adenoma composed of small tubules in a myxoid background. |
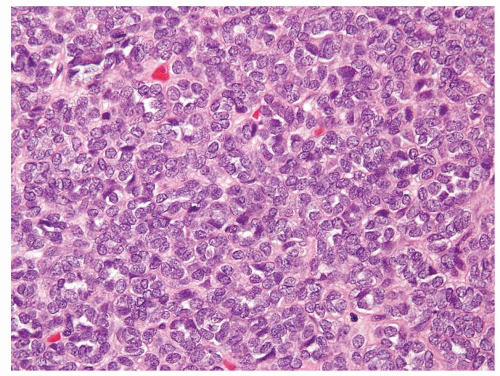 Figure 2.2.5 Solid area of metanephric adenoma that is composed of uniform small nuclei without mitoses. |
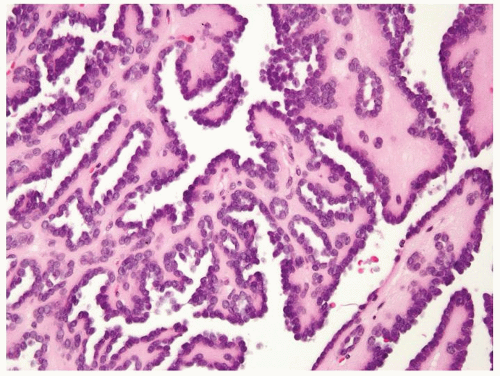 Figure 2.2.6 Metanephric adenoma with papillary morphology. |
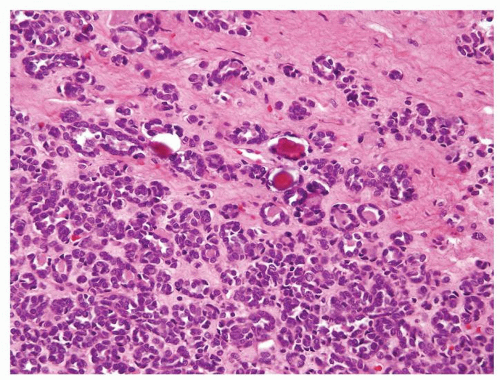 Figure 2.2.7 Metanephric adenoma with calcifications. |
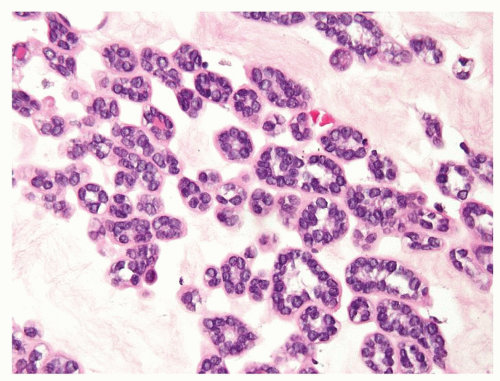 Figure 2.2.8 Metanephric adenoma with myxoid stroma. |
|
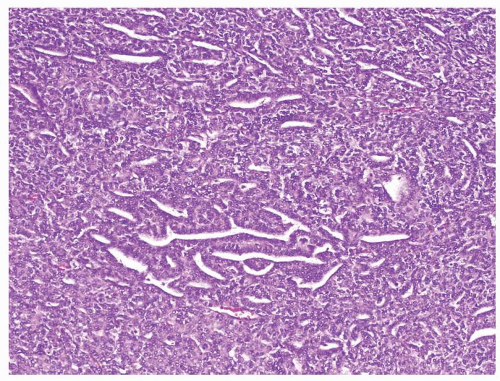 Figure 2.3.1 Wilms tumor with favorable histology. Nuclear pleomorphism is lacking even at a low magnification. |
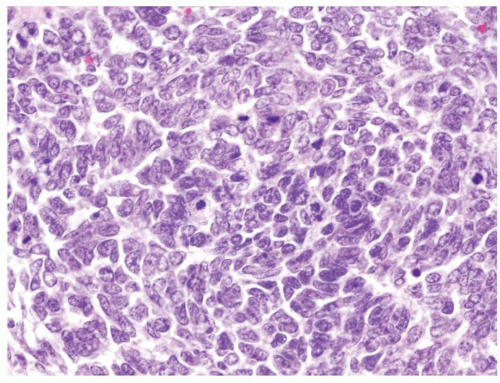 Figure 2.3.2 Wilms tumor with favorable histology. Abnormal mitotic figures are not found. Nuclear pleomorphism is lacking. |
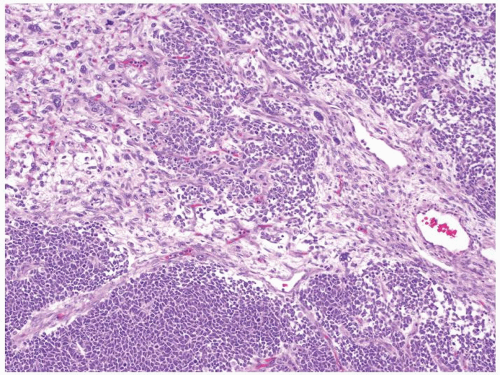 Figure 2.3.3 Wilms tumor with unfavorable histology. “Anaplastic nuclear change” is the hallmark of unfavorable histology; markedly enlarged hyperchromatic nuclei seen on 10× objective. |
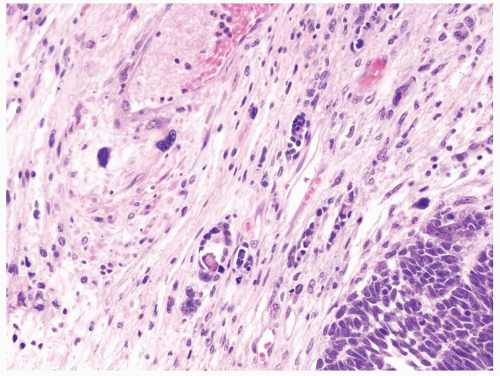 Figure 2.3.4 Wilms tumor with unfavorable histology with markedly enlarged hyperchromatic nuclei (upper left) compared to favorable histology in lower right. |
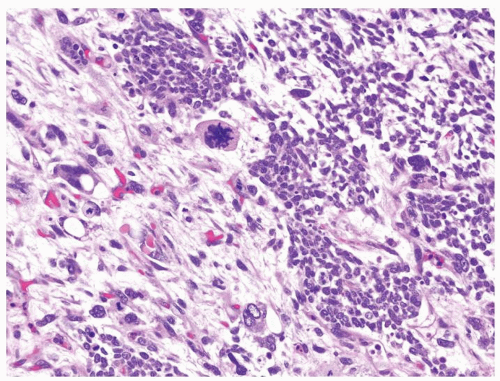 Figure 2.3.5 Wilms tumor with unfavorable histology. Polyploidy/multipolar mitotic figures. |
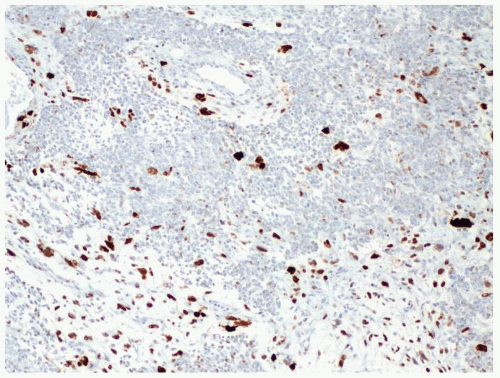 Figure 2.3.6 Wilms tumor with unfavorable histology. p53 positivity in areas of anaplastic nuclear change. |
|
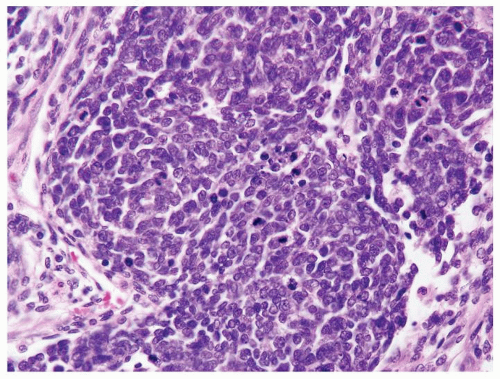 Figure 2.4.1 Wilms tumor with blastema showing hyperchromatic overlapping nuclei. |
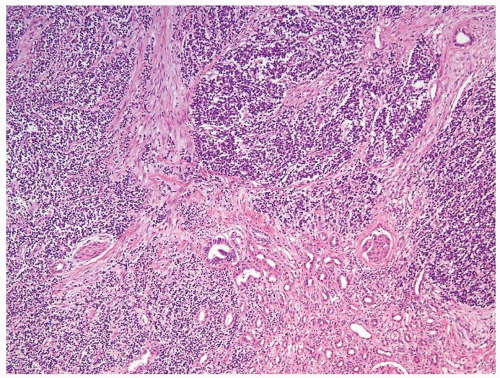 Figure 2.4.2 PNET/Ewing tumor involving the kidney. |
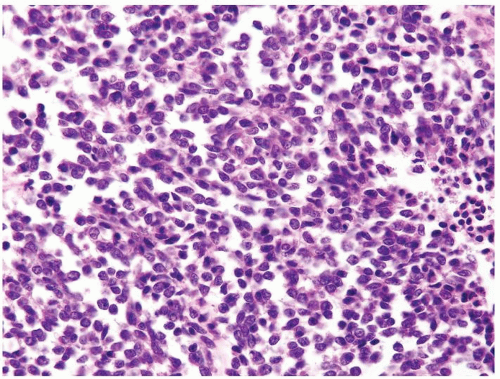 Figure 2.4.3 Same case as Figure 2.4.2 with monotonous polygonal cells with thin rim of cleared cytoplasm. Less hyperchromatic nuclei than WT blastemal cells and more evenly spaced. |
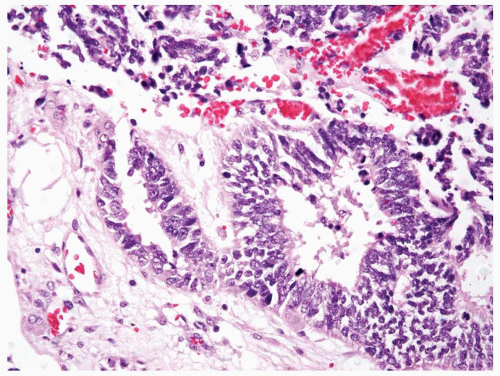 Figure 2.4.4 Renal PNET/Ewing tumors arranged in Homer-Wright rosettes. |
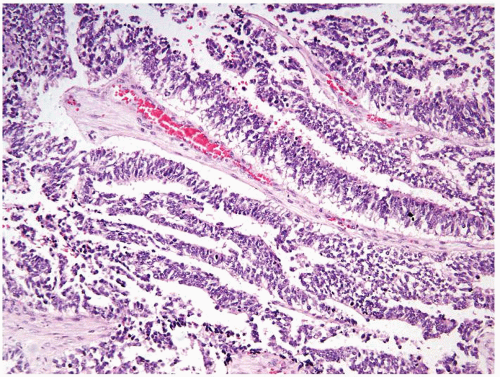 Figure 2.4.5 Same case as Figure 2.4.4 with perivascular pseudorosettes. |
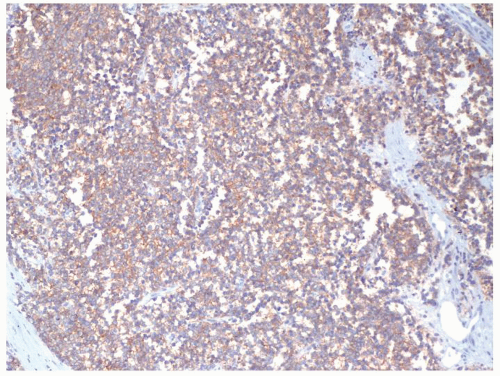 Figure 2.4.6 Same case as Figures 2.4.4 and 2.4.5 with diffuse membranous CD99 positivity. |
|
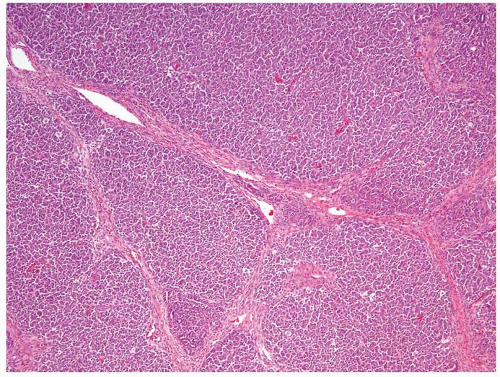 Figure 2.5.1 Wilms tumor with a nodular pattern. |
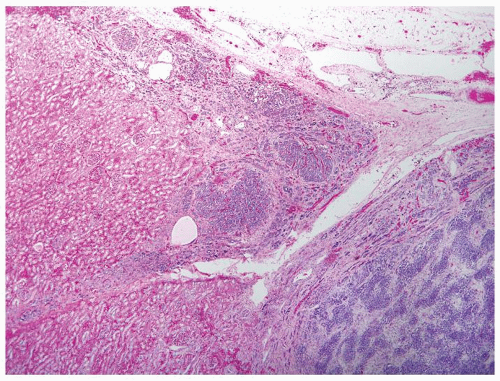 Figure 2.5.2 Nephrogenic rest (center and upper left) adjacent to serpentine pattern of Wilms tumor (lower right). |
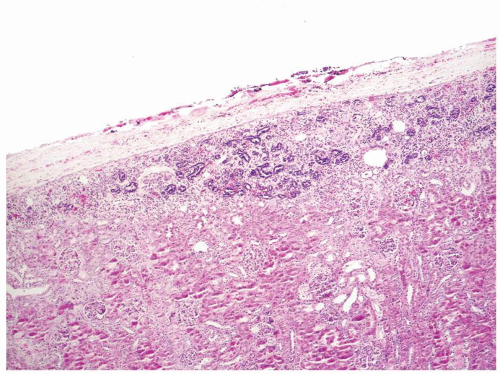 Figure 2.5.3 Perilobar sclerosing nephrogenic rest. |
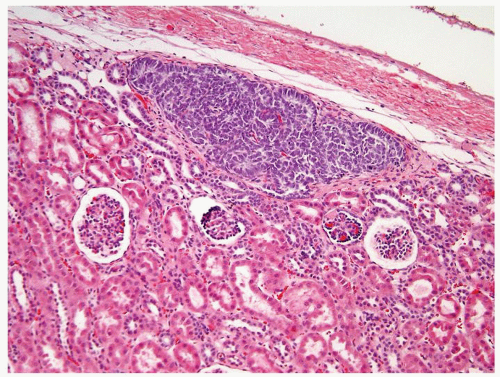 Figure 2.5.4 Perilobar nephrogenic rest. (Courtesy of Dr. Peter Argani.) |
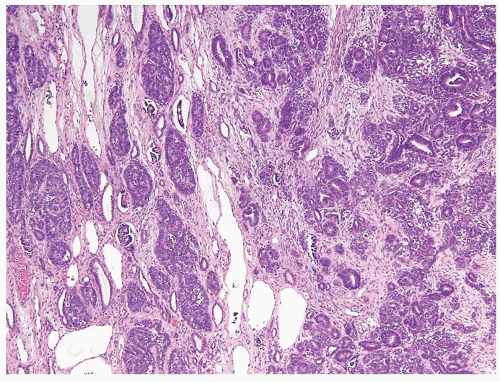 Figure 2.5.5 Intralobar rest admixed with normal kidney (left) adjacent to solid Wilms tumor (right). (Courtesy of Dr. Peter Argani.) |
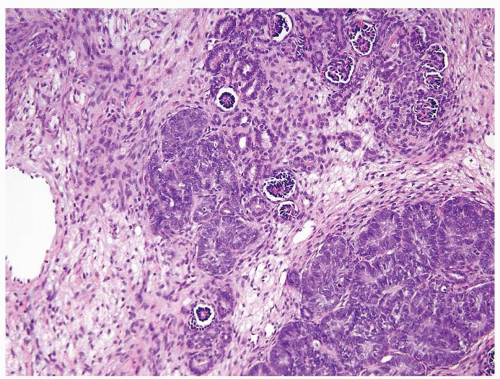 Figure 2.5.6 Same case as Figure 2.5.5 with intralobar rest and associated stroma admixed with a normal kidney. |
|
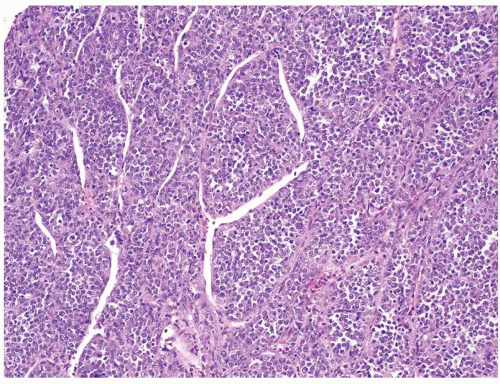 Figure 2.6.1 Rhabdoid tumor of the kidney with discohesive sheets of undifferentiated tumor. |
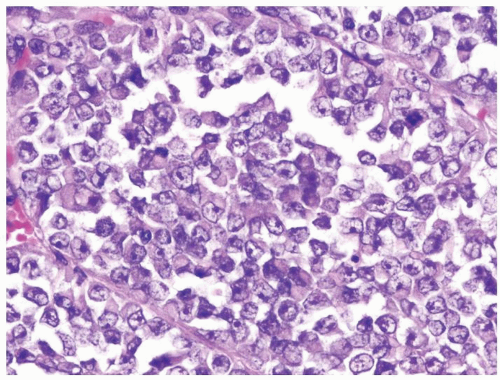 Figure 2.6.2 Same case as Figure 2.6.1 with polygonal, large-sized cells with abundant cytoplasm and eccentric nuclei containing prominent nucleoli. |
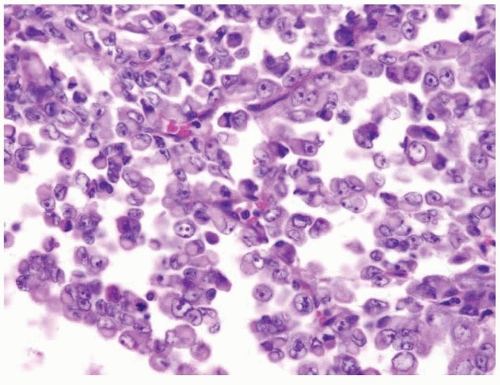 Figure 2.6.3 Rhabdoid tumor of the kidney. |
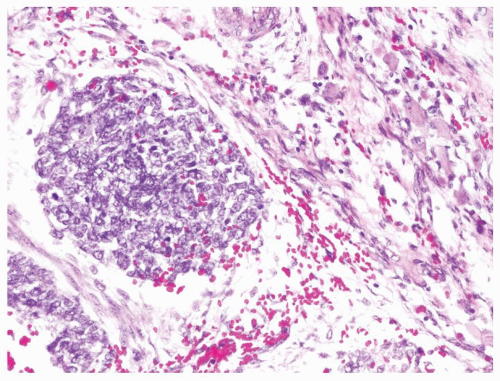 Figure 2.6.4 Wilms tumor with blastema and rhabdoid cells. |
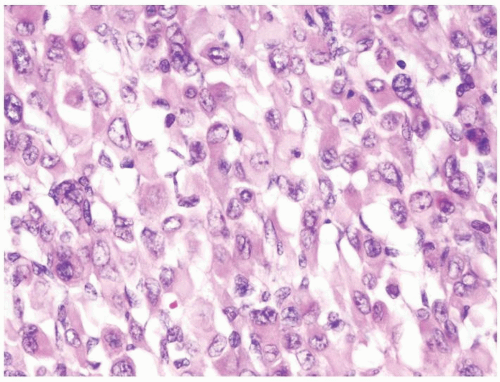 Figure 2.6.5 Same case as Figure 2.6.3 with rhabdoid differentiation in stromal component. |
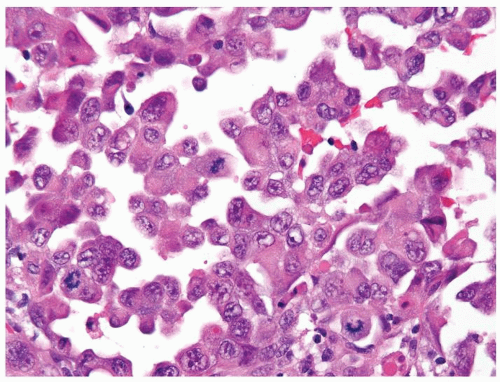 Figure 2.6.6 Medullary carcinoma of the kidney with rhabdoid features. |
|
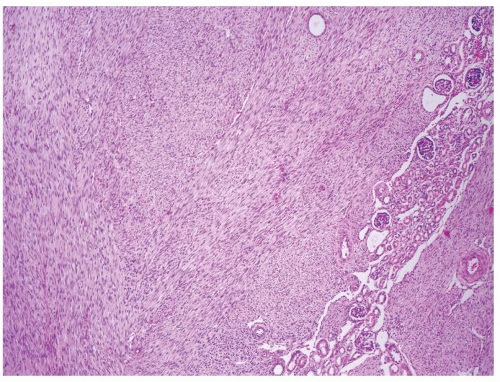 Figure 2.7.1 Mesoblastic nephroma extending into surrounding the kidney. |
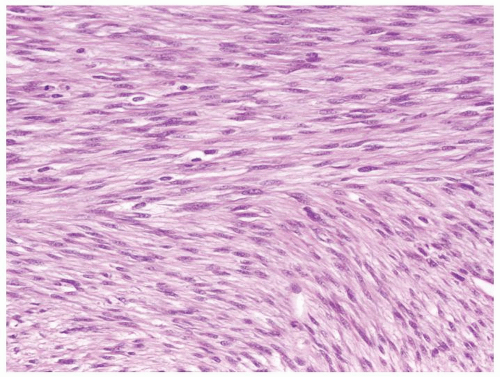 Figure 2.7.2 Same case as Figure 2.7.1 with monomorphous spindle fibroblastic- and myofibroblastic-type cells. |
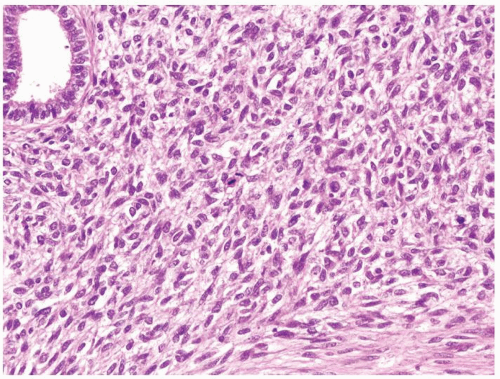 Figure 2.7.3 Cellular mesoblastic nephroma with moderate pleomorphism, greater cellularity, and increased mitotic activity. |
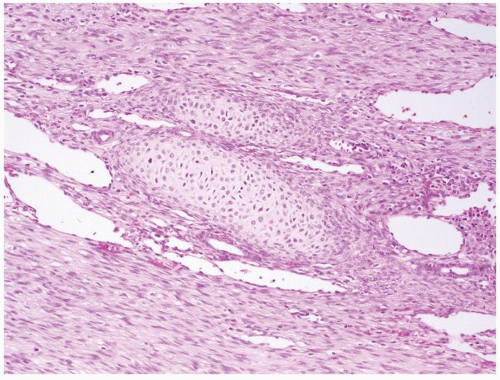 Figure 2.7.4 Mesoblastic nephroma with nodules of hyaline cartilage. |
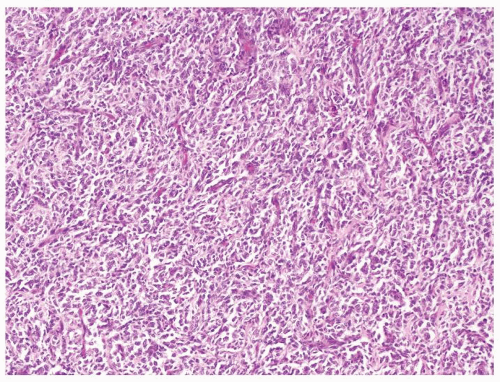 Figure 2.7.5 Clear cell sarcoma with chicken-wire vascular pattern. |
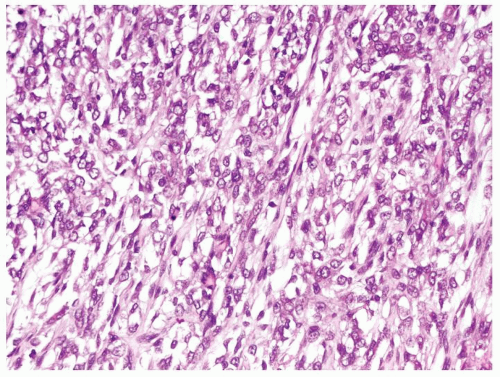 Figure 2.7.6 Same case as Figure 2.7.5 with monomorphous spindle to epithelioid cells loosely set in extracellular myxoid material. |
|
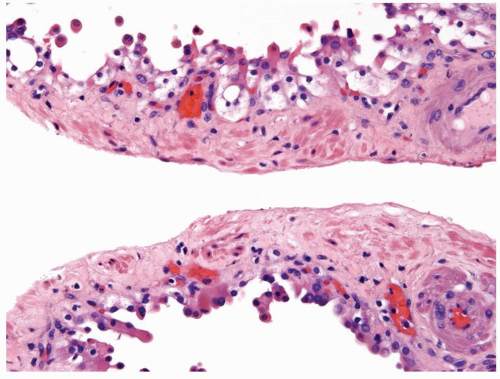 Figure 2.8.1 Atypical cyst with piled up clear cells but lacking clear cells within the wall of the septae. |
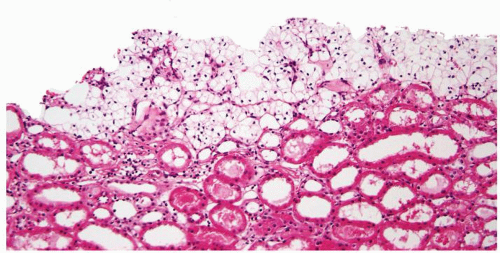 Figure 2.8.2 Atypical cyst piled up clear cells with abortive papillae. |
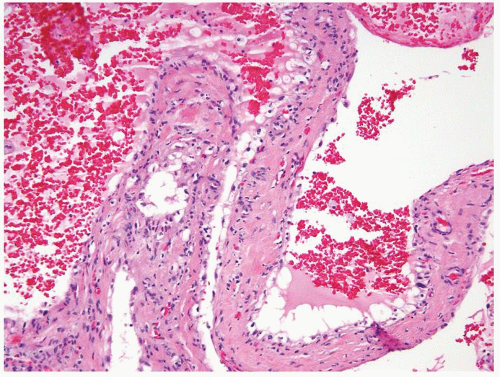 Figure 2.8.3 Simple cyst lined by a single layer of clear cells. |
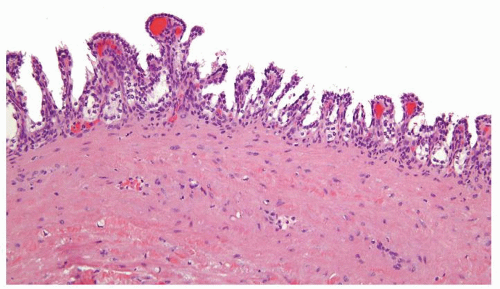 Figure 2.8.4 Atypical cyst with papillary projections lined by cuboidal epithelium with clear cytoplasm. |
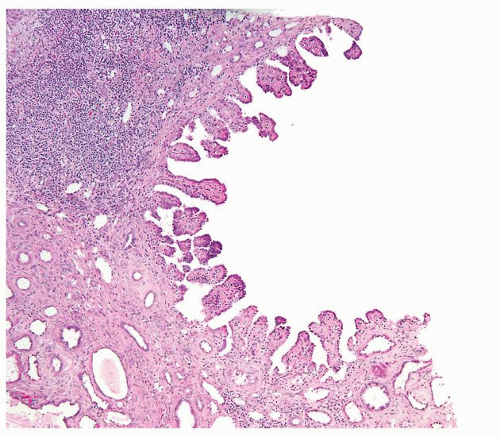 Figure 2.8.5 Unilocular cyst with short papillary projections into the cyst. This lesion is incapable of metastatic behavior and should not be designated as carcinoma. |
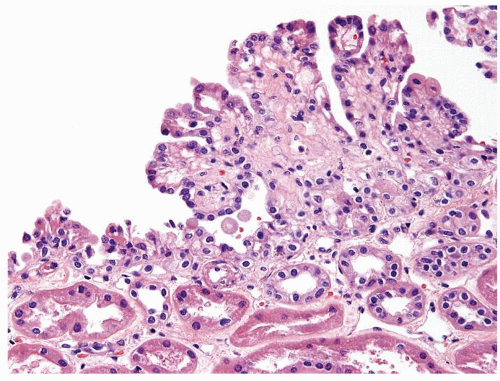 Figure 2.8.6 Unilocular cyst with minute focus of short papillary projections lined by bland cuboidal cells. This lesion is incapable of metastatic behavior and should not be designated as carcinoma. |
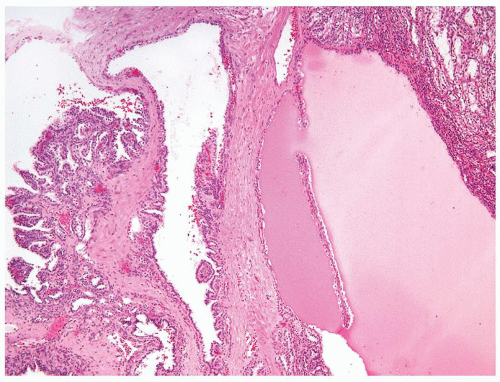 Figure 2.8.7 Cystic papillary RCC with complex papillary nodule (left) and nodule of tubules lined by clear cells (upper right). |
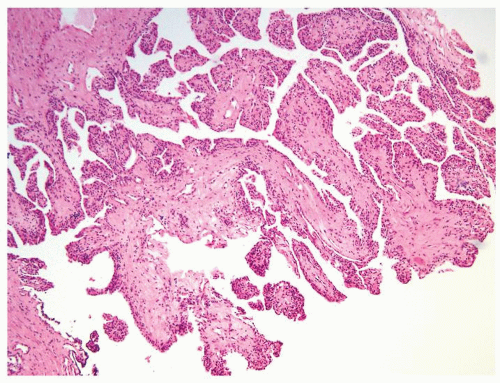 Figure 2.8.8 Cystic papillary RCC. |
|
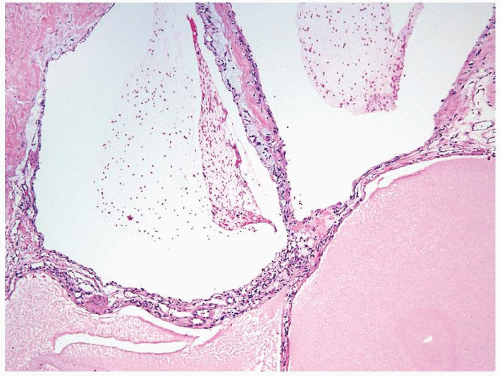 Figure 2.9.1 Multicystic renal cell neoplasm of low malignant potential (LMP) showing variable-sized cysts lined by cuboidal clear cells. |
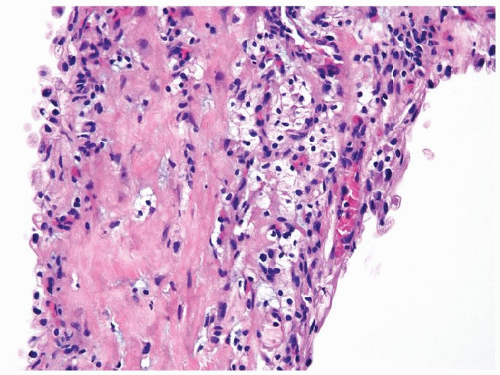 Figure 2.9.2 Same case as Figure 2.9.1 with septae containing rare small nonexpansile aggregates of cells with clear cytoplasm and round-to-ovoid nuclei. |
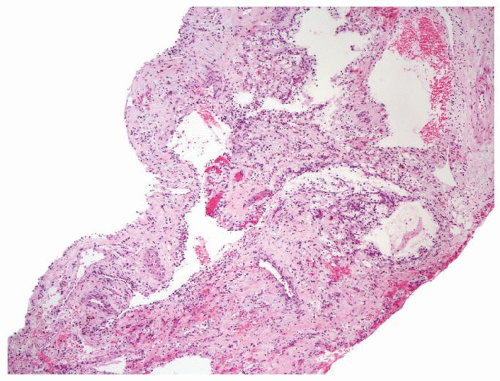 Figure 2.9.3 Multicystic renal cell neoplasm of low malignant potential (LMP) without obvious carcinoma at low magnification. |
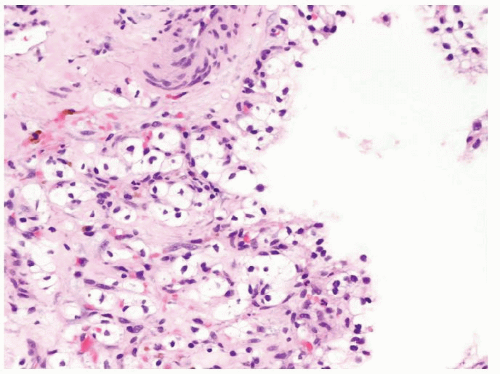 Figure 2.9.4 Same case as Figure 2.9.3 with cytologically benign nonexpansile clear cells within septae. |
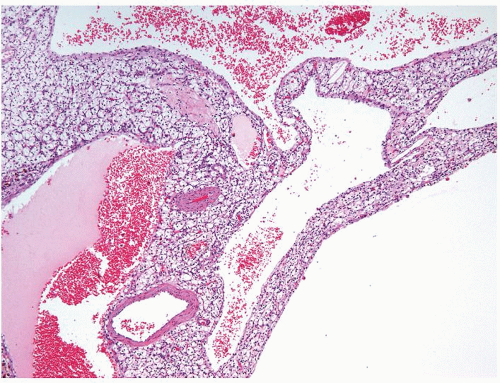 Figure 2.9.5 Cystic clear cell renal cell carcinoma showing variable-sized cysts lined by cuboidal clear cells. Septa contain expansile solid tumor. |
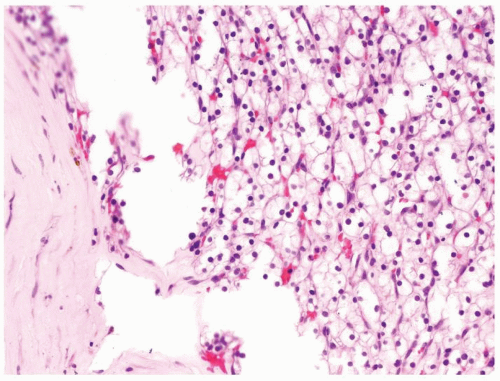 Figure 2.9.6 Same case as Figure 2.9.5 with solid nests of clear cells. |
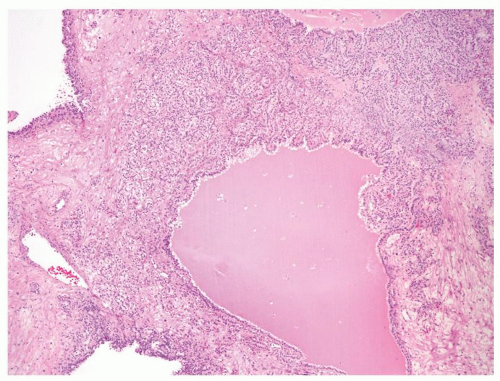 Figure 2.9.7 Cystic clear cell renal cell carcinoma with septae containing expansile solid of nests of tumor. |
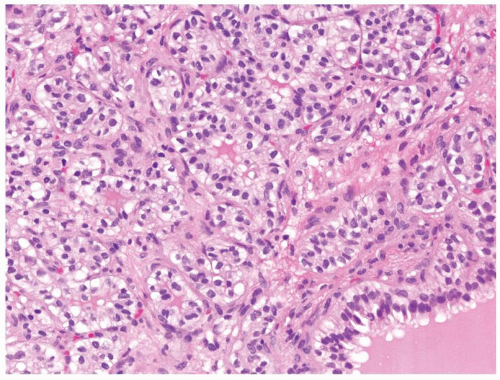 Figure 2.9.8 Same case as Figure 2.9.7 with low-grade renal cell carcinoma within septae. |
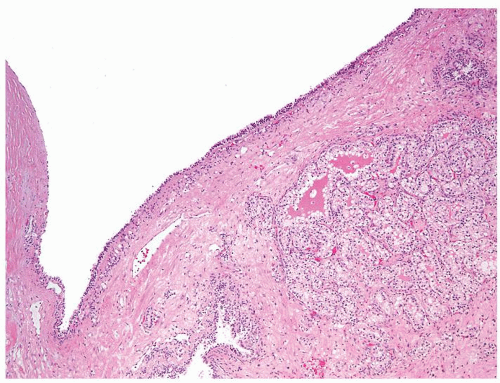 Figure 2.9.9 Cystic clear cell renal cell carcinoma with nodule of carcinoma within wall. |
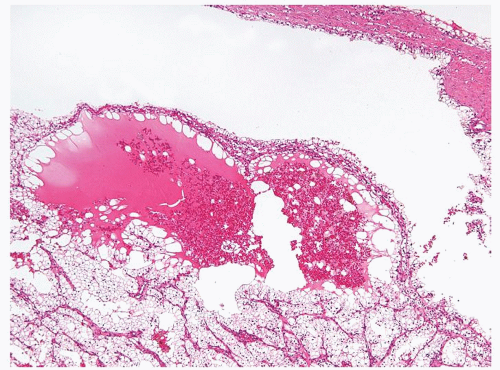 Figure 2.9.10 Cystic clear cell renal cell carcinoma with nodule of carcinoma within wall. |
|
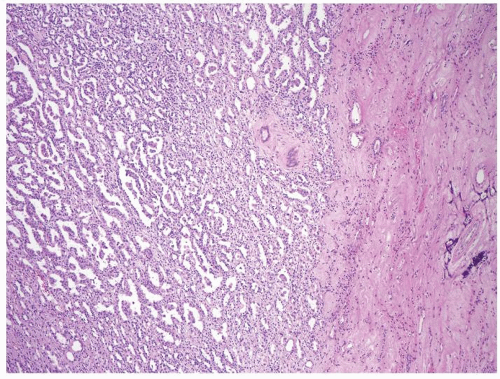 Figure 2.10.1 Solid papillary renal cell carcinoma with paler appearance at low magnification. |
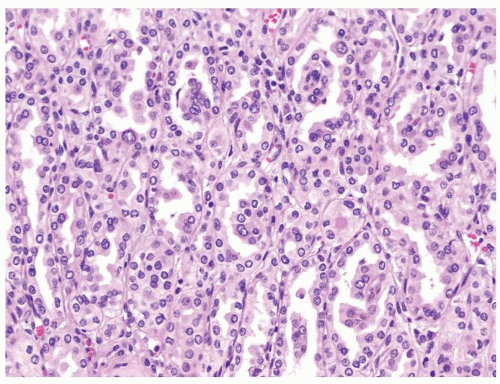 Figure 2.10.2 Same case as Figure 2.10.1 composed of stubby glomeruloid papillary structures lacking well-formed fibrovascular cores admixed with tubular proliferation. Epithelial cells with moderate amount of eosinophilic cytoplasm. |
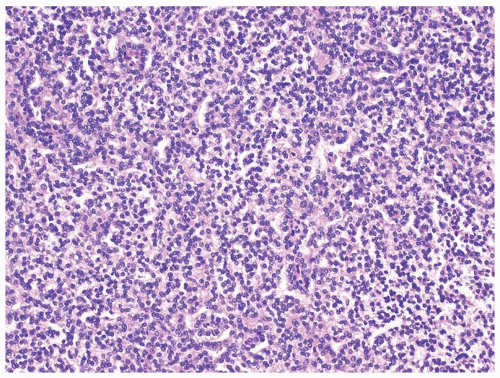 Figure 2.10.3 Solid papillary renal cell carcinoma with a more compact appearance. |
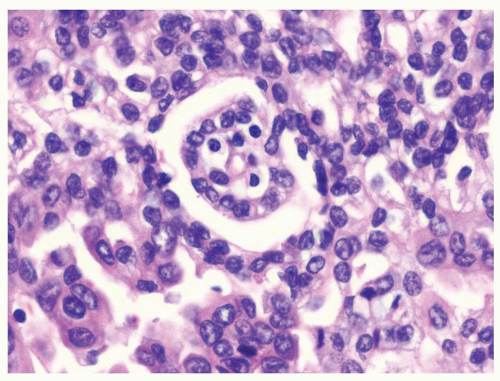 Figure 2.10.4 Same case as Figure 2.10.3 with occasional stubby glomeruloid papillary structures. |
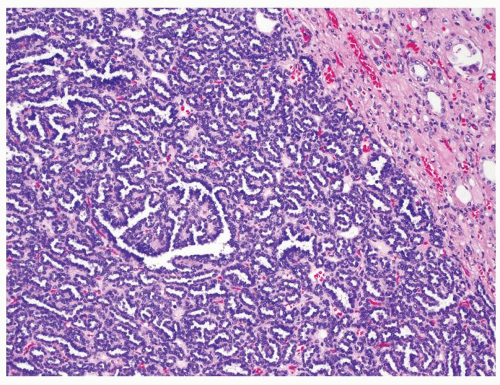 Figure 2.10.5 Metanephric adenoma showing “blue/basophilic” low-power appearance. |
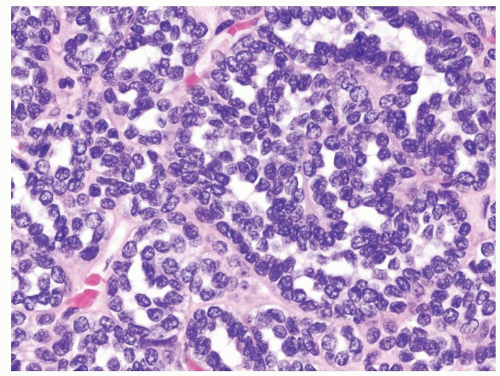 Figure 2.10.6 Same case as Figure 2.10.5 showing a very high N/C ratio with bland cytology. |
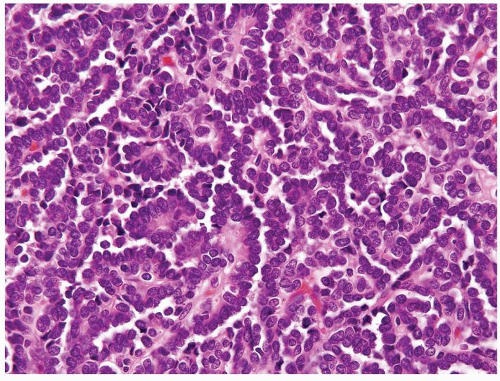 Figure 2.10.7 Metanephric adenoma with papillary formation. |
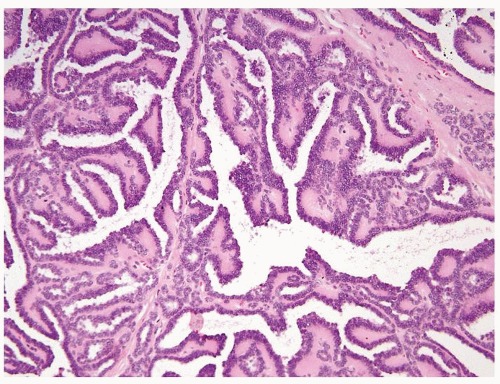 Figure 2.10.8 Metanephric adenoma with papillary formation. Note more typical small tubules in the wall. |
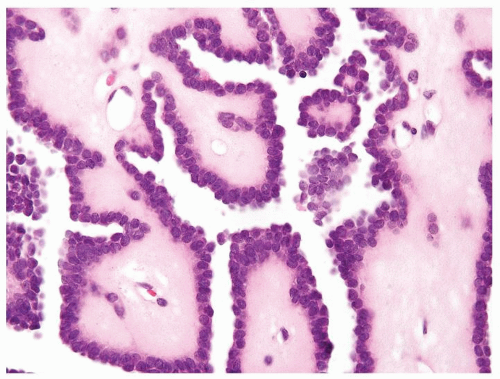 Figure 2.10.9 Same case as Figure 2.10.8 with bland cells with a high N/C ratio in myxoid stroma typical of metanephric adenoma. |
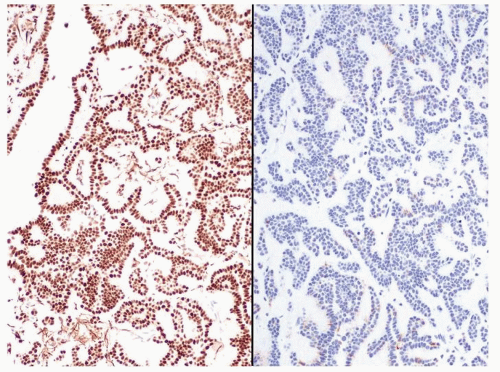 Figure 2.10.10 Metanephric adenoma positive for WT-1 (left) and negative for CK7 (right). Solid papillary RCC has the opposite immunohistochemical profile. |
|
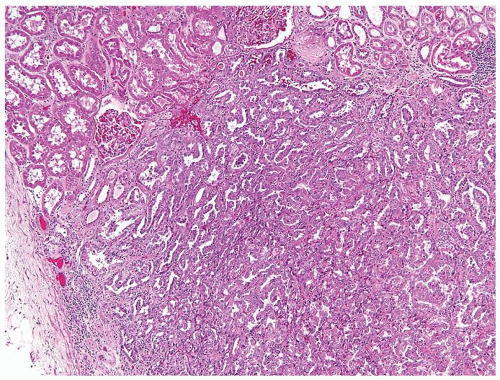 Figure 2.11.1 Papillary renal adenoma. Size of the lesion (<5 mm) differentiates from carcinoma as well as the lack of a capsule and nuclear atypia. |
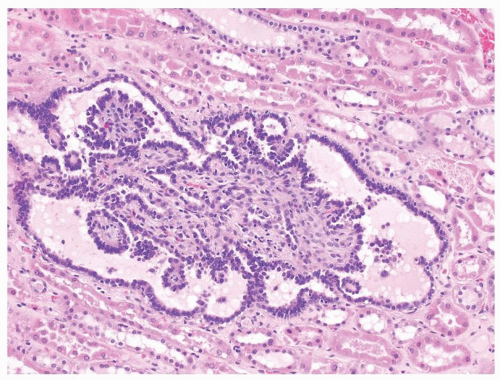 Figure 2.11.2 Tubulopapillary renal hyperplasia with a single dilated tubule containing papillary projections. The lesion is nonexpansile. |
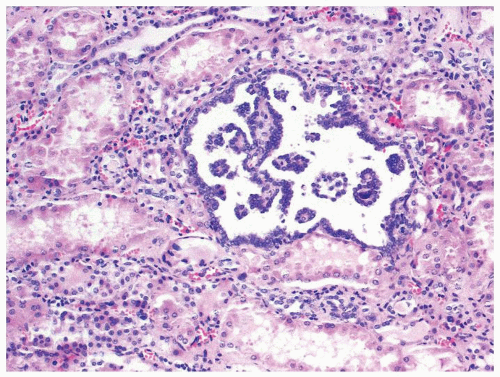 Figure 2.11.3 Tubulopapillary renal hyperplasia. |
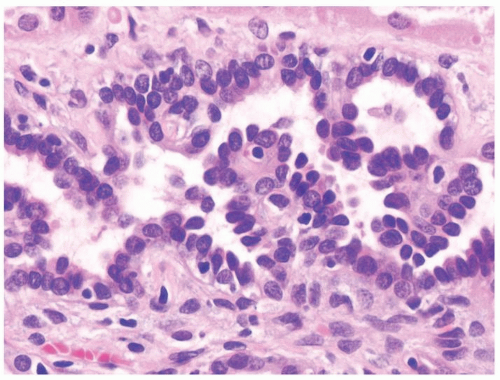 Figure 2.11.4 Tubulopapillary renal hyperplasia with bland cytology. |
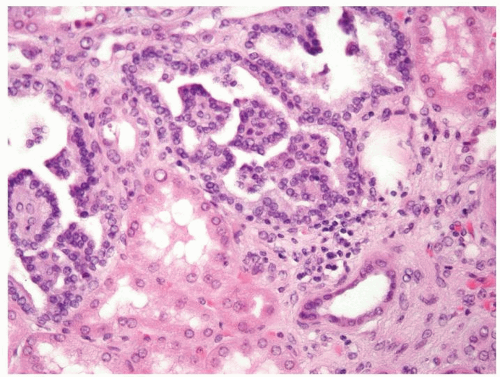 Figure 2.11.5 Tubulopapillary renal hyperplasia.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|