OBJECTIVES
After studying this chapter, you should be able to:
List the passages through which air passes from the exterior to the alveoli, and describe the cells that line each of them.
List the major muscles involved in respiration, and state the role of each.
Define the basic measures of lung volume and give approximate values for each in a normal adult.
Define lung compliance and airway resistance.
Compare the pulmonary and systemic circulations, and list some major differences between them.
Describe basic lung defense and metabolic functions.
Define partial pressure and calculate the partial pressure of each of the important gases in the atmosphere at sea level.
INTRODUCTION
The structure of the respiratory system is uniquely suited to its primary function, the transport of gases in and out of the body. In addition, the respiratory system provides a large volume of tissue that is constantly exposed to the outside environment, and thus, to potential infection and injury. Finally, the pulmonary system includes a unique circulation that must handle the blood flow. This chapter begins with the basic anatomy and cellular physiology that contribute to the respiratory system and some of their unique features. The chapter also includes discussion of how the anatomic features contribute to the basic mechanics of breathing, as well as some highlights of nonrespiratory physiology in the pulmonary system.
ANATOMY OF THE LUNGS
Airflow through the respiratory system can be broken down into three interconnected regions: the upper airway; the conducting airway; and the alveolar airway (also known as the lung parenchyma or acinar tissue). The upper airway consists of the entry systems, the nose/nasal cavity and mouth that lead into the pharynx. The larynx extends from the lower part of the pharynx to complete the upper airway. The nose is the primary point of entry for inhaled air; therefore, the mucosal epithelium lining the nasopharyngeal airways is exposed to the highest concentration of inhaled allergens, toxicants, and particulate matter. With this in mind, it is easy to understand that in addition to olfaction, the nose and upper airway provides two additional crucial functions in airflow—(1) filtering out large particulates to prevent them from reaching the conducting and alveolar airways and (2) serving to warm and humidify air as it enters the body. Particulates larger than 30–50 μm in size tend to not to be inhaled through the nose whereas particulates on the order of 5–10 μm impact on the nasopharynx and do enter the conducting airway. Most of these latter particles settle on mucous membranes in the nose and pharynx. Because of their momentum, they do not follow the airstream as it curves downward into the lungs, and they impact on or near the tonsils and adenoids, large collections of immunologically active lymphoid tissue in the back of the pharynx.
The conducting airway begins at the trachea and branches dichotomously to greatly expand the surface area of the tissue in the lung. The first 16 generations of passages form the conducting zone of the airways that transports gas from and to the upper airway described above (Figure 34–1). These branches are made up of bronchi, bronchioles, and terminal bronchioles. The conducting airway is made up of a variety of specialized cells that provide more than simply a conduit for air to reach the lung (Figure 34–2). The mucosal epithelium is attached to a thin basement membrane, and beneath this, the lamina propria. Collectively these are referred to as the “airway mucosa.” Smooth muscle cells are found beneath the epithelium and an enveloping connective tissue is likewise interspersed with cartilage that is more predominant in the portions of the conducting airway of greater caliber. The epithelium is organized as a pseudostratified epithelium and contains several cell types, including ciliated and secretory cells (eg, goblet cells and glandular acini) that provide key components for airway innate immunity, and basal cells that can serve as progenitor cells during injury. As the conducting airway transitions to terminal and transitional bronchioles, the histologic appearance of the conducting tubes change. Secretory glands are absent from the epithelium of the bronchioles and terminal bronchioles, smooth muscle plays a more prominent role and cartilage is largely absent from the underlying tissue. Club cells (formerly termed “Clara cells”), nonciliated cuboidal epithelial cells that secrete important defense markers and serve as progenitor cells after injury, make up a large portion of the epithelial lining in the latter portions of the conducting airway.
FIGURE 34–1
Conducting and respiratory zones in the airway. A) Resin cast of the human airway tree shows dichotomous branching beginning at the trachea. Note the added pulmonary arteries (red) and veins (blue) displayed in the left lung. B) The branching pattern of the airway is sketched with individual regions of the conducting and respiratory airways identified. (Reproduced with permission from Fishman AP; Fishman’s Pulmonary Disease and Disorders, 4th ed. New York, NY: McGraw Hill Medical; 2008.)


FIGURE 34–2
Cellular transition from conducting airway to the alveolus. The epithelial layer transitions from pseudostratified layer with submucosal glands to a cuboidal epithelium and then to a squamous epithelium. The underlying mesynchyme tissue and capillary structure also changes with the airway transition. (Adapted with permission from Fishman AP: Fishman’s Pulmonary Diseases and Disorders, 4th ed. New York, NY: McGraw Hill Medical; 2008.)
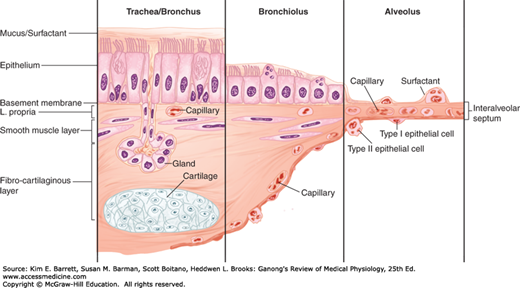
Epithelial cells in the conducting airway can secrete a variety of molecules that aid in lung defense. Secretory immunoglobulins (IgA), collectins (including surfactant protein (SP) -A and SP-D), defensins and other peptides and proteases, reactive oxygen species, and reactive nitrogen species are all generated by airway epithelial cells. These secretions can act directly as antimicrobials to help keep the airway free of infection. Airway epithelial cells also secrete a variety of chemokines and cytokines that recruit traditional immune cells and other immune effector cells to site of infections. The smaller particles that make it through the upper airway, ~2–5 μm in diameter, generally fall on the walls of the bronchi as the airflow slows in the smaller passages. There they can initiate reflex bronchial constriction and coughing. Alternatively, they can be moved away from the lungs by the “mucociliary escalator.” The epithelium of the respiratory passages from the anterior third of the nose to the beginning of the respiratory bronchioles is ciliated (Figure 34–2). The cilia are bathed in a periciliary fluid where they typically beat at rates of 10–15 Hz. On top of the periciliary layer and the beating cilia rests a mucus layer, a complex mixture of proteins and polysaccharides secreted from specialized cells, glands, or both in the conducting airway. This combination allows for the trapping of foreign particles (in the mucus) and their transport out of the airway (powered by ciliary beat). The ciliary mechanism is capable of moving particles away from the lungs at a rate of at least 16 mm/min. When ciliary motility is defective, as can occur in smokers, or as a result of other environmental conditions or genetic deficiencies, mucus transport is virtually absent. This can lead to chronic sinusitis, recurrent lung infections, and bronchiectasis. Some of these symptoms are evident in cystic fibrosis (Clinical Box 34–1).
The walls of the bronchi and bronchioles are innervated by the autonomic nervous system. Nerve cells in the airways sense mechanical stimuli or the presence of unwanted substances in the airways such as inhaled dusts, cold air, noxious gases, and cigarette smoke. These neurons can signal the respiratory centers to contract the respiratory muscles and initiate sneeze or cough reflexes. The receptors show rapid adaptation when they are continuously stimulated to limit sneeze and cough under normal conditions. The β2-adrenergic receptors help mediate bronchodilation. They also increase bronchial secretions (eg, mucus), while α1-adrenergic receptors inhibit secretions.
CLINICAL BOX 34–1 Cystic Fibrosis
Among whites, cystic fibrosis is one of the most common genetic disorders; greater than 3% of the United States population are carriers for this autosomal recessive disease.
The gene that is abnormal in cystic fibrosis is located on the long arm of chromosome 7 and encodes the cystic fibrosis transmembrane conductance regulator (CFTR), a regulated Cl– channel located on the apical membrane of various secretory and absorptive epithelia. The number of reported mutations in the CFTR gene that cause cystic fibrosis is large (> 1000) and the mutations are now grouped into five classes (I–V) based on their cellular function. Class I mutations do not allow for synthesis of the protein. Class II mutations have protein processing defects. Class III mutations have a block in their channel regulation. Class IV mutations display altered conductance of the ion channel. Class V mutations display reduced synthesis of the protein. The severity of the defect varies with the class and the individual mutation. The most common mutation causing cystic fibrosis is loss of the phenylalanine residue at amino acid position 508 of the protein (ΔF508), a Class II mutation that limits the amount of protein that gets to the plasma membrane.
One outcome of cystic fibrosis is repeated pulmonary infections, particularly with Pseudomonas aeruginosa, and progressive, eventually fatal destruction of the lungs. There is also suppressed chloride secretion across the wall of the airways. One would expect Na+ reabsorption to be depressed as well, and indeed in sweat glands it is. However, in the lungs, it is enhanced, so that the Na+ and water move out of airways, leaving their other secretions inspissated and sticky. This results in a reduced periciliary layer that inhibits function of the mucociliary escalator, and alters the local environment to reduce the effectiveness of antimicrobial secretions.
THERAPEUTIC HIGHLIGHTSTraditional treatments of cystic fibrosis address the various symptoms. Chest physiotherapy and mucolytics are used to loosen thick mucus and aid lung clearance. Antibiotics are used to prevent new infections and keep chronic infections in check. Bronchodilators and anti-inflammatory medications are used to help expand and clear air passages. Pancreatic enzymes and nutritive supplements are used to increase nutrient absorption and promote weight gain. Because of the “single gene” mutation of this disease, gene therapy has been closely examined; however, results have not been successful. More recently, drugs that target the molecular defects have been advancing in clinical trials and are showing great promise for better treatments.
Between the trachea and the alveolar sacs, the airways divide 23 times. The last seven generations form the transitional and respiratory zones where gas exchange occurs are made up of transitional and respiratory bronchioles, alveolar ducts, and alveoli (Figure 34–1). These multiple divisions greatly increase the total cross-sectional area of the airways, from 2.5 cm2 in the trachea to 11,800 cm2 in the alveoli. Consequently, the velocity of airflow in the small airways declines to very low values. The transition from the conducting to the respiratory region that ends in the alveoli also includes a change in cellular arrangements (Figure 34–2; and Figure 34–3). Humans have 300 million alveoli, and the total area of the alveolar walls in contact with capillaries in both lungs is about 70 m2.
FIGURE 34–3
Prominent cells in the adult human alveolus. A) A cross-section of the respiratory zone shows the relationship between capillaries and the airway epithelium. Only 4 of the 18 alveoli are labeled. B) Enlargement of the boxed area from (A) displaying intimate relationship between capillaries, the interstitium, and the alveolar epithelium. C) Electron micrograph displaying a typical area depicted in (B). The pulmonary capillary (cap) in the septum contains plasma with red blood cells. Note the closely apposed endothelial and pulmonary epithelial cell membranes separated at places by additional connective tissue fibers (cf); en, nucleus of endothelial cell; epl, nucleus of type I alveolar epithelial cell; a, alveolar space; ma, alveolar macrophage. D) Type II cell formation and metabolism of surfactant. Lamellar bodies (LB) are formed in type II alveolar epithelial cells and secreted by exocytosis into the fluid lining the alveoli. The released lamellar body material is converted to tubular myelin (TM), and the TM is the source of the phospholipid surface film (SF). Surfactant is taken up by endocytosis into alveolar macrophages and type II epithelial cells. N, nucleus; RER, rough endoplasmic reticulum; Golgi, Golgi apparatus; CB, composite body. (For (A) Reproduced with permission from Greep RO, Weiss L: Histology, 3rd ed. New York: New York, NY: McGraw-Hill; 1973; (B) Adapted with permission from Gong H, Drage CW: The Respiratory System: a core curriculum. Norwalk, CT: Appleton-Century-Crofts: 1982; (C) Reproduced with permission from Burri PA: Development and growth of the human lung. In: Handbook of Physiology, Section 3, The Respiratory System. Fishman AP, Fisher AB [editors]. American Physiological Society, 1985; and (D) Reproduced with permission from Wright JR: Metabolism and turnover of lung surfactant. Am Rev Respir Dis Aug; 136(2):426–444, 1987.)

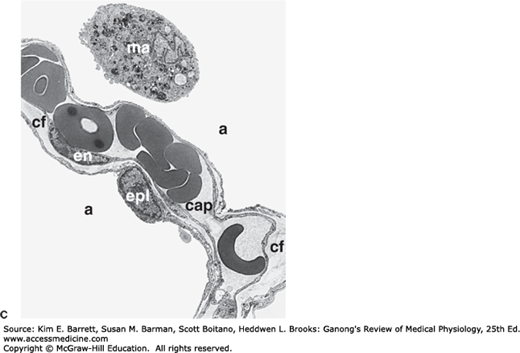

The alveoli are lined by two types of epithelial cells. Type I cells are flat cells with large cytoplasmic extensions and are the primary lining cells of the alveoli, covering approximately 95% of the alveolar epithelial surface area. Type II cells (granular pneumocytes) are thicker and contain numerous lamellar inclusion bodies. Although these cells make up only 5% of the surface area, they represent approximately 60% of the epithelial cells in the alveoli. Type II cells are important in alveolar repair as well as other cellular physiology. One prime function of the type II cell is the production of surfactant (Figure 34–3D). Typical lamellar bodies, membrane-bound organelles containing whorls of phospholipid, are formed in these cells and secreted into the alveolar lumen by exocytosis. Tubes of lipid called tubular myelin form from the extruded bodies, and the tubular myelin in turn forms a phospholipid film. Following secretion, the phospholipids of surfactant line up in the alveoli with their hydrophobic fatty acid tails facing the alveolar lumen. This surfactant layer plays an important role in maintaining alveolar structure by reducing surface tension (see below). Surface tension is inversely proportional to the surfactant concentration per unit area. The surfactant molecules move further apart as the alveoli enlarge during inspiration, and surface tension increases, whereas it decreases when they move closer together during expiration. Some of the protein-lipid complexes in surfactant are taken up by endocytosis in type II alveolar cells and recycled.
The alveoli are surrounded by pulmonary capillaries. In most areas, air and blood are separated only by the alveolar epithelium and the capillary endothelium, so they are about 0.5 μm apart (Figure 34–3). The alveoli also contain other specialized cells, including pulmonary alveolar macrophages (PAMs, or AMs), lymphocytes, plasma cells, neuroendocrine cells, and mast cells. PAMs are an important component of the pulmonary defense system. Like other macrophages, these cells come originally from the bone marrow. PAMs are actively phagocytic and ingest small particles that evade the mucociliary escalator and reach the alveoli. They also help process inhaled antigens for immunologic attack, and they secrete substances that attract granulocytes to the lungs as well as substances that stimulate granulocyte and monocyte formation in the bone marrow. PAM function can also be detrimental—when they ingest large amounts of the substances in cigarette smoke or other irritants, they may release lysosomal products into the extracellular space to cause inflammation.
The lungs are positioned within the thoracic cavity, which is defined by the rib cage and the spinal column. The lungs are surrounded by a variety of muscles that contribute to breathing (Figure 34–4). Movement of the diaphragm accounts for 75% of the change in intrathoracic volume during quiet inspiration. Attached around the bottom of the thoracic cage, this muscle arches over the liver and moves downward like a piston when it contracts. The distance it moves ranges from 1.5 cm to as much as 7 cm with deep inspiration.
FIGURE 34–4
Muscles and movement in respiration. A) An idealized diagram of respiratory muscles surrounding the rib cage. The diaphragm and intercostals play prominent roles in respiration. B) and C) Radiograph of chest in full expiration (B) and full inspiration (C). The dashed white line in C is an outline of the lungs in full expiration. Note the difference in intrathoracic volume. (Reproduced with permission (A) Reproduced with permission from Fishman AP: Fishman’s Pulmonary Diseases and Disorders, 4th ed. New York, NY: McGraw Hill Medical; 2008; (B, C) Reproduced with permission from Comroe JH Jr: Physiology of Respiration, 2nd ed., Year Book; 1975.)
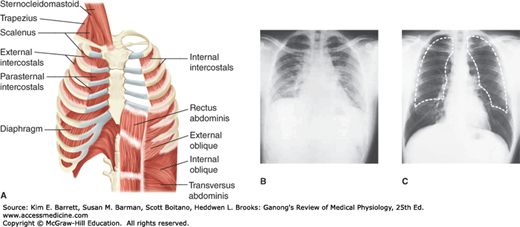
The diaphragm has three parts: the costal portion, made up of muscle fibers that are attached to the ribs around the bottom of the thoracic cage; the crural portion, made up of fibers that are attached to the ligaments along the vertebrae; and the central tendon, into which the costal and the crural fibers insert. The central tendon is also the inferior part of the pericardium. The crural fibers pass on either side of the esophagus and can compress it when they contract. The costal and crural portions are innervated by different parts of the phrenic nerve and can contract separately. For example, during vomiting and eructation, intra-abdominal pressure is increased by contraction of the costal fibers but the crural fibers remain relaxed, allowing material to pass from the stomach into the esophagus.
The other important inspiratory muscles are the external intercostal muscles, which run obliquely downward and forward from rib to rib. The ribs pivot as if hinged at the back, so that when the external intercostals contract they elevate the lower ribs. This pushes the sternum outward and increases the anteroposterior diameter of the chest. The transverse diameter also increases but to a lesser degree. Either the diaphragm or the external intercostal muscles alone can maintain adequate ventilation at rest. Transection of the spinal cord above the third cervical segment is fatal without artificial respiration, but transection below the fifth cervical segment is not because it leaves the phrenic nerves that innervate the diaphragm intact; the phrenic nerves arise from cervical segments 3–5. Conversely, in patients with bilateral phrenic nerve palsy but intact innervation of their intercostal muscles, respiration is somewhat labored but adequate to maintain life. The scalene and sternocleidomastoid muscles in the neck are accessory inspiratory muscles that help elevate the thoracic cage during deep labored respiration.
A decrease in intrathoracic volume and forced expiration result when the expiratory muscles contract. The internal intercostals have this action because they pass obliquely downward and posteriorly from rib to rib and therefore pull the rib cage downward when they contract. Contractions of the muscles of the anterior abdominal wall also aid expiration by pulling the rib cage downward and inward and by increasing the intra-abdominal pressure, which pushes the diaphragm upward.
In order for air to get into the conducting airway it must pass through the glottis, defined as the area including and between the vocal folds within the larynx. The abductor muscles in the larynx contract early in inspiration, pulling the vocal cords apart and opening the glottis. During swallowing or gagging, a reflex contraction of the adductor muscles closes the glottis and prevents aspiration of food, fluid, or vomitus into the lungs. In unconscious or anesthetized patients, glottic closure may be incomplete and vomitus may enter the trachea, causing an inflammatory reaction in the lung (aspiration pneumonia).
The pleural cavity and its infoldings serve as a lubricating fluid/area that allows for lung movement within the thoracic cavity (Figure 34–5A). There are two layers that contribute to the pleural cavity: the parietal pleura and the visceral pleura. The parietal pleura is a membrane that lines the chest cavity containing the lungs. The visceral pleura is a membrane that lines the lung surface. The pleural fluid (~15–20 mL) forms a thin layer between the pleural membranes and prevents friction between surfaces during inspiration and expiration.
FIGURE 34–5
Pleural space and connective fibers. A) Front sectional drawing of lung within the rib cage. Note the parietal and visceral pleura and the infoldings around the lung lobes that include pleural space. B) Connective fiber tracts of the lung are highlighted. Note the axial fibers along the airways, the peripheral fibers in the pleura, and the septal fibers. (Adapted with permission from Fishman AP: Fishman’s Pulmonary Diseases and Disorders, 4th ed. New York, NY: McGraw Hill Medical; 2008.)
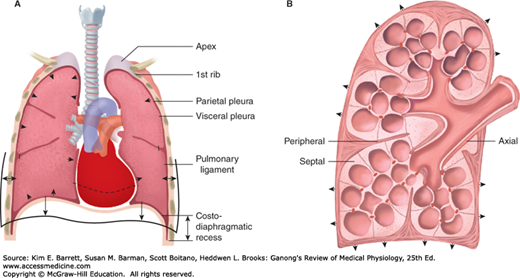
The lung itself contains a vast amount of free space—it is ~80% air. Although this maximizes surface area for gas exchange, it also requires an extensive support network to maintain lung shape and function. The connective tissue within the visceral pleura contains three layers that help support the lung. Elastic fibers that follow the mesothelium effectively wrap the three lobes of the right lung and the two lobes of the left lung (Figure 34–5B). A deep sheet of fine fibers that follow the outline of the alveoli provide support to individual air sacks. Between these two separate sheets lies connective tissue that is interspersed with individual cells for support and lung maintenance/function.
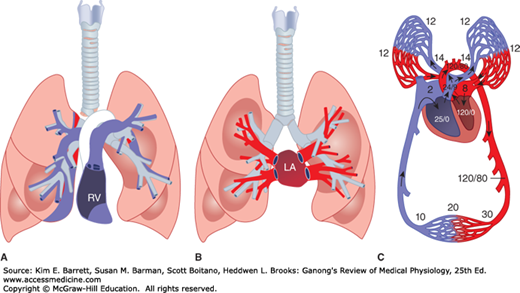
Both the pulmonary circulation and the bronchial circulation contribute to blood flow in the lung. In the pulmonary circulation (Figure 34–6), almost all the blood in the body passes via the pulmonary artery to the pulmonary capillary bed, where it is oxygenated and returned to the left atrium via the pulmonary veins. The pulmonary arteries strictly follow the branching of the bronchi down to the respiratory bronchioles. The pulmonary veins, however, are spaced between the bronchi on their return to the heart. The separate and much smaller bronchial circulation includes the bronchial arteries that come from systemic arteries. They form capillaries, which drain into bronchial veins or anastomose with pulmonary capillaries or veins. The bronchial veins drain into the azygos vein. The bronchial circulation nourishes the trachea down to the terminal bronchioles and also supplies the pleura and hilar lymph nodes. It should be noted that lymphatic channels are more abundant in the lungs than in any other organ. Lymph nodes are arranged along the bronchial tree and extend down until the bronchi are ~5 mm in diameter. Lymph node sizes can range from 1 mm at the bronchial periphery to 10 mm along the trachea. The nodes are connected by lymph vessels and allow for unidirectional flow of lymph to the subclavian veins.
FIGURE 34–6
Pulmonary circulation. A) Schematic diagram of the relation of the main branches of the pulmonary arteries to the bronchial tree. B) Schematic of the pulmonary veins in relation to the bronchial tree. C) Representative areas of blood flow are labeled with corresponding blood pressure (mm Hg). In all diagrams, blue represents de-oxygenated blood and red represents oxygenated blood. The deoxygenated/oxygenated shift in C occurs as blood traverses capillaries that wrap individual alveoli and is not as abrupt as shown. LA, left atrium; RV, right ventricle. (For (A, B) Reproduced with permission from Fishman AP: Fishman’s Pulmonary Diseases and Disorders, 4th ed. New York, NY: McGraw Hill Medical; 2008; (C) Modified with permission from Comroe JH Jr: Physiology of Respiration, 2nd ed. Year Book; 1974.)
MECHANICS OF RESPIRATION
The lungs and the chest wall are elastic structures. Normally, no more than a thin layer of fluid is present between the lungs and the chest wall (intrapleural space). The lungs slide easily on the chest wall, but resist being pulled away from it in the same way that two moist pieces of glass slide on each other but resist separation. The pressure in the “space” between the lungs and chest wall (intrapleural pressure) is subatmospheric (Figure 34–7). The lungs are stretched when they expand at birth, and at the end of quiet expiration their tendency to recoil from the chest wall is just balanced by the tendency of the chest wall to recoil in the opposite direction. If the chest wall is opened, the lungs collapse; and if the lungs lose their elasticity, the chest expands and becomes barrel-shaped.
FIGURE 34–7
Pressure in the alveoli and the pleural space relative to atmospheric pressure during inspiration and expiration. The dashed line indicates what the intrapleural pressure would be in the absence of airway and tissue resistance; the actual curve (solid line) is skewed to the left by the resistance. Volume of breath during inspiration/expiration is graphed for comparison.
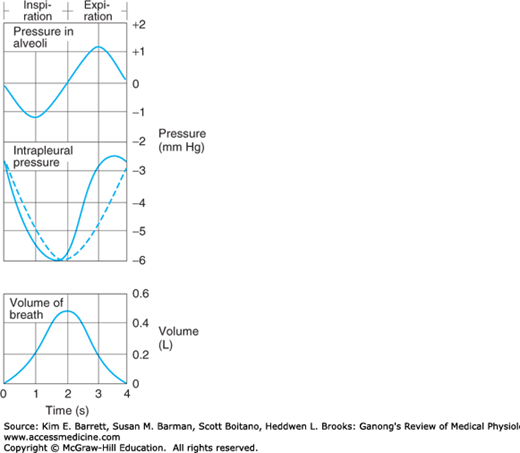
Inspiration is an active process. The contraction of the inspiratory muscles increases intrathoracic volume. The intrapleural pressure at the base of the lungs, which is normally about –2.5 mm Hg (relative to atmospheric) at the start of inspiration, decreases to about –6 mm Hg. The lungs are pulled into a more expanded position. The pressure in the airway becomes slightly negative, and air flows into the lungs. At the end of inspiration, the lung recoil begins to pull the chest back to the expiratory position, where the recoil pressures of the lungs and chest wall balance (see below). The pressure in the airway becomes slightly positive, and air flows out of the lungs. Expiration during quiet breathing is passive in the sense that no muscles that decrease intrathoracic volume contract. However, some contraction of the inspiratory muscles occurs in the early part of expiration. This contraction exerts a braking action on the recoil forces and slows expiration. Strong inspiratory efforts reduce intrapleural pressure to values as low as –30 mm Hg, producing correspondingly greater degrees of lung inflation. When ventilation is increased, the extent of lung deflation is also increased by active contraction of expiratory muscles that decrease intrathoracic volume.
Modern spirometers permit direct measurement of gas intake and output. Since gas volumes vary with temperature and pressure and since the amount of water vapor in them varies, these devices have the ability to correct respiratory measurements involving volume to a stated set of standard conditions. It should be noted that correct measurements are highly dependent on the ability for the practitioner to properly encourage the patient to fully utilize the device. Modern techniques for gas analysis make possible rapid, reliable measurements of the composition of gas mixtures and the gas content of body fluids. For example, O2 and CO2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


