Inhibition of the Gastric Acid Pump
H+ transport by the gastric ATPase is the final step in acid secretion. It follows that inhibition of the ATPase would be an effective means of regulating acid secretion and cannot be surmounted by alteration of stimulatory pathways. There are various parameters that need to be considered when designing a drug for inhibition of this process by inhibiting the ATPase.
There has to be a large therapeutic index. The acid related diseases are in general not life threatening, and, although theoretically less effective, there are several H2 receptor antagonists available, both as generics and OTC in most countries in the world. These latter drugs have a large margin of safety, and compounds that treat acid related diseases by inhibition of the gastric acid pump have to have at least an equal margin of safety. There are several P-type ATPases, and a gastric drug needs to be selective for the gastric ATPase. The ATPase is a member of the P2-type ATPase family, with homology to the Na,K ATPases and the colonic H,K ATPase. Hence, inhibitors of the H,K ATPase must not interfere with functioning of other ATPases and also should affect as few other targets as possible.
Whereas design of small-ligand antagonists is not easy, at least the structure of the natural ligand provides a template for such molecules. In the case of the ATPase, the natural ligands are ATP and the hydronium ion, H3O+, on the cytoplasmic side and K+ on the luminal surface. A compound competing with ATP in cell cytoplasm would obviously be nonspecific, inhibiting all other ATP-dependent processes. Design of molecules to act as H3O+ surrogates would have to take advantage of a special structure in the pump able to bind hydronium ion without affecting other such binding sites. Similar considerations would apply to design of compounds substituting for K+ on the outside surface. Clearly, such compounds could not be designed based on the structure of the transported cations.
An advantage accrues in drug design from the specialized location of the functioning ATPase. In the actively secreting parietal cell, the active pumps are present in the microvilli of the secretory canaliculus and produce a pH gradient by far the most acidic space in the body. It is approximately 1,000-fold more acidic than anywhere else. Protonatable weak bases will accumulate selectively in the canalicular space as a function of their pKa. This is because the protonated positively charged form is significantly less permeable than the uncharged species. Because this is the only space in the body with a pH of less than 4.0, weak bases with a pKa of 4.0 or less will accumulate only in the canaliculus of active parietal cells. The unprotonated form will permeate the basal-lateral and canalicular membrane of the parietal cell; the protonated form, being relatively membrane impermeable, will concentrate in the canaliculus. In the fully active cell, the pH of the canalicular lumen is approximately 1.0; therefore, a compound with a pKa of 4.0 will accumulate approximately 1,000-fold in this space and gain a significant therapeutic advantage if presented to the parietal cell in the serosal space. The acidity of the space could
also confer a chemical advantage if such weak bases are inactive but are also acid labile. These would then convert more rapidly in the acidic secretory canaliculus to compounds able to inhibit the ATPase. As will be seen, this prodrug concept has played a vital role in the development of the current drugs used to control acid secretion by covalent pump inhibition.
also confer a chemical advantage if such weak bases are inactive but are also acid labile. These would then convert more rapidly in the acidic secretory canaliculus to compounds able to inhibit the ATPase. As will be seen, this prodrug concept has played a vital role in the development of the current drugs used to control acid secretion by covalent pump inhibition.
P2-type ATPases
There are several mammalian P ATPases, and each has several isoforms, but there are sufficient differences in amino acid sequence to enable specificity of inhibitory agents. For example, the cardioglycosides, such as digoxin, have only one known target, the Na,K ATPase. Similarly, thapsigargin targets the sarcoplasmic and endoplasmic reticular but not the plasma membrane Ca ATPase. Bafilomycin targets only the V-type ATPases and oligomycin, largely the F1F0 ATPase. There is, therefore, good reason to expect that drugs targeted against the gastric H,K ATPase will have strict specificity if properly designed.
Targets of the PPIs
The three-dimensional structure of the gastric acid pump is based on the structure of the SR Ca ATPase, and there is sufficient difference in detail that this structure, although highly informative as to mechanism, cannot provide a template for design of specific inhibitory ligands. Design of the PPIs does not depend on details of molecular structure, because these are chemical in concept, rather than biologic.
It is possible to define the amino acids in proteins that are able to react under biologic conditions. Whereas relatively harsh conditions are used for chemical reactions with carboxylic acids, histidines, lysines, arginines, and tyrosines, the most reactive amino acids under biologic conditions are cysteines by virtue of the higher reactivity of the −SH group. Given that an −SH-reactive group has to be generated in the canalicular space, it is the cysteines accessible from the luminal space that are potential specific targets for thiolreactive (thiophilic) prodrugs. Obviously, the conversion of the prodrug to the cysteine-reactive thiophilic compound should occur predominantly in the acidic-secretory canaliculus of the active parietal cell. If it were formed elsewhere, rapid reaction with other proteins and glutathione would be found, inactivating the drug and possibly causing side effects.
It is, therefore, pertinent to inquire as to which of the 28 cysteines in the catalytic subunit of the pump and which of the nine cysteines of the beta subunit are available for stable derivatization. From the analysis of the structure of the membrane domain of the catalytic subunit, there are at least five possible cysteines that are accessible from the luminal side of the pump to a fairly bulky cationic organic molecule: the cysteines at positions 321, 813, 822, 892, and 981 in or between the third, fifth, and sixth membrane segments, in the loop between TM7 and TM8, and at the luminal end of TM9, respectively. Because the cysteines in the beta subunit are in disulfide linkage, either they are not attacked or they reform after reaction.
The substituted pyridal methylsulfinyl benzimidazoles
Discovery of PPIs
The development of the first of this series of drugs is due to a combination of serendipity, analysis of mechanism, and conviction that the ATPase was the best target for control of acid secretion. A compound, pyridine-2-acetamide, had been purchased by a company (Hässle in Göteborg, Sweden) for possible use as an antiviral agent. This compound was found ineffective but had some antisecretory activity. It was modified to pyridine-2-thioacetamide to improve its antiviral efficacy, but this did not happen, although it retained its antisecretory activity. In 1973, SK&F announced the development of cimetidine, the world’s first H2 receptor antagonist. Based on the structure of cimetidine, a benzimidazole ring was added to the antisecretory pyridine-2-thioacetamide, with the hope that the mechanism of action of these forerunners was H2 antagonism. Antisecretory activity was retained. Finally, the sulfide was modified for stabilization to a sulfoxide, and timoprazole was born.
This compound had rather remarkable antisecretory properties: it inhibited gastric acid secretion, whatever the stimulus; it inhibited secretion in isolated gastric glands, whatever the stimulus, but was relatively acid unstable and showed inhibition of iodide uptake by the thyroid and was thymotoxic.
The first polyclonal antibody against the H,K ATPase reacted with the stomach, of course, but also mysteriously with the thyroid and thymus. This suggested that perhaps the ATPase was the target of timoprazole. By 1977, a
compound, picoprazole, had been made, retaining the core structure of timoprazole. It was shown that this compound and timoprazole did inhibit the gastric ATPase only when the ATPase was making acid, and that there was a lag phase of inhibition of transport activity. Because the compound was a weak base, the steps that were thought to result in inhibition of ATPase activity and acid secretion involved accumulation of the compound in the acid space of the isolated gastric vesicle during H+ transport (or the parietal cell canaliculus), followed by acid-dependent conversion to an active compound. It was postulated that this class of compound acted as a prodrug that only reacted with the ATPase after this acid catalyzed conversion to an active form, perhaps the sulfenic acid. Later, it was proven that the active form in solution was a rearranged planar tetracyclic compound containing a highly reactive sulfenamide group. To optimize the acid stability of the parent compound and to generate absolute selectivity for accumulation in the acid space of the parietal cell, omeprazole was synthesized in 1979 and became the compound that was launched in 1988 at the Rome World Congress of Gastroenterology.
compound, picoprazole, had been made, retaining the core structure of timoprazole. It was shown that this compound and timoprazole did inhibit the gastric ATPase only when the ATPase was making acid, and that there was a lag phase of inhibition of transport activity. Because the compound was a weak base, the steps that were thought to result in inhibition of ATPase activity and acid secretion involved accumulation of the compound in the acid space of the isolated gastric vesicle during H+ transport (or the parietal cell canaliculus), followed by acid-dependent conversion to an active compound. It was postulated that this class of compound acted as a prodrug that only reacted with the ATPase after this acid catalyzed conversion to an active form, perhaps the sulfenic acid. Later, it was proven that the active form in solution was a rearranged planar tetracyclic compound containing a highly reactive sulfenamide group. To optimize the acid stability of the parent compound and to generate absolute selectivity for accumulation in the acid space of the parietal cell, omeprazole was synthesized in 1979 and became the compound that was launched in 1988 at the Rome World Congress of Gastroenterology.
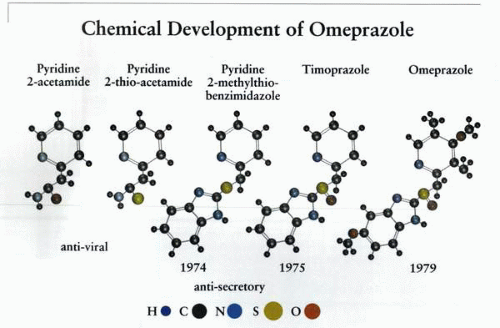 The gradual generation of compounds able to inhibit acid secretion. The last two—timoprazole and omeprazole—are PPIs. The mechanism of action of the first two compounds of the series are probably as K+-competitive antagonists, and the middle compound is probably oxidized to timoprazole in the liver. (H, hydrogen; C, carbon; N, nitrogen; S, sulfur; O, oxygen.) |
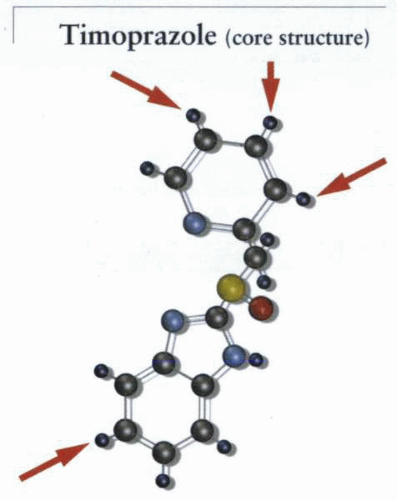 The core structure of all the clinically approved PPIs is timoprazole. Although effective as an acid-suppressive agent, this molecule was unstable at neutral pH, as well as exhibited thyrotoxic and thymotoxic effects. This led to the development of similar chemical moieties that were not only stable at neutral pH, but also exhibited no organ toxicity. The arrows show the position of substitution of different groups to produce the different PPIs now on the market. The initial substitutions designed for omeprazole resulted in the loss of the side effects of timoprazole. |
The name coined for this class of drug was proton pump inhibitor (PPI). After publication in 1981 of the first of a series of papers on the mechanism of action of these drugs, a variety of derivatives were synthesized that also led to the introduction of other drugs with generally similar properties to omeprazole.
The core structure of all currently marketed benzimidazoles, with points of substitution, is shown in the figure.
Mechanism of action
Chemistry
The key steps are (a) acid gradient-dependent accumulation of the prodrugs and (b) an acid- or enzyme surfacecatalyzed conversion of the prodrugs to a sulfenic acid and then to a tetracyclic, planar sulfenamide. A detailed mechanistic analysis of the chemistry behind the substituted benzimidazoles was published recently. Recent findings may suggest that the pump itself may play a role in its own inhibition in that perhaps the sulfenic acid is formed on the surface of the pump, and this is the reactive intermediate binding to the relevant cysteines.
The structure of four examples of these drugs is given in the illustration. It can be seen that they all share the same backbone, a 2-pyridyl methylsulfinyl benzimidazole with various substitutions on the pyridine or benzimidazole moieties, which are added to modify solution reactivity or to decrease toxicity.
Design criteria
1. pKa
Biologic accumulation of weakly basic compounds depends on a pH gradient between solution or cytoplasm and a membrane-bounded or enclosed compartment
of lower pH. The protonated form of the weak base is relatively membrane impermeable, and, therefore, the chemical accumulates in acidic compartments, depending on the gradient and the pKa of the chemical. There are various acidic compartments, such as lysosomes, neurosecretory granules, and endosomes, that have a pHi of approximately 4.5 to 5.0. The active secretory canaliculus of a fully stimulated parietal cell has a pHni of close to 0.8. Therefore, to target only the acidic compartment of the parietal cell, the pKa of a PPI must be 4.5 or less to avoid accumulation in lysosomes or neurosecretory granules. The accumulation of these drugs preferentially in the acid space of the secreting parietal cell gives them an excellent therapeutic index, or margin of safety.
of lower pH. The protonated form of the weak base is relatively membrane impermeable, and, therefore, the chemical accumulates in acidic compartments, depending on the gradient and the pKa of the chemical. There are various acidic compartments, such as lysosomes, neurosecretory granules, and endosomes, that have a pHi of approximately 4.5 to 5.0. The active secretory canaliculus of a fully stimulated parietal cell has a pHni of close to 0.8. Therefore, to target only the acidic compartment of the parietal cell, the pKa of a PPI must be 4.5 or less to avoid accumulation in lysosomes or neurosecretory granules. The accumulation of these drugs preferentially in the acid space of the secreting parietal cell gives them an excellent therapeutic index, or margin of safety.
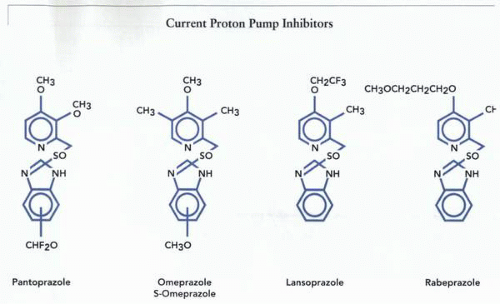 The current proton pump inhibitors, pantoprazole, omeprazole, lansoprazole, and rabeprazole. The 2D structure of esomeprazole is the same as omeprazole, but the 3D structure differs in the orientation of the S→O group. |
2. Membrane permeability
Because the compound accesses this compartment by crossing both the plasma membrane and the canalicular membrane, it also has to be membrane permeable, requiring a log P of between -3 and 0. Too high a log P drives the chemical into the hydrophobic domain of the membrane bilayer, perhaps decreasing access to the target or generating an inappropriate target site; too low a log P reduces membrane permeability and access to the acid space.
3. Acid activation
The prodrug forms, as shown, are ampholytic, with three N atoms able to accept or donate protons. The important site of protonation for accumulation of these drugs is the pyridine N, with a pKa of 4.0 or thereabout. This pKa ensures that accumulation of the prodrug will occur only in the secreting parietal cell canaliculus. Acid activation depends on protonation of the benzimidazole N followed by reaction of the 2C with the unprotonated pyridine N.
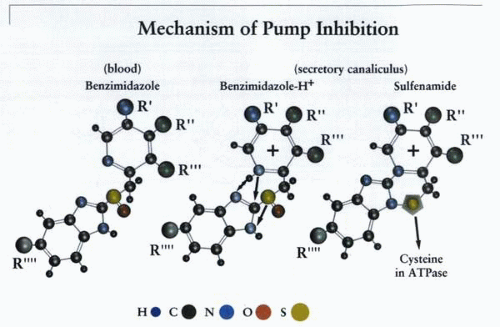 Mechanism of action of the PPIs, showing acidity-dependent accumulation, conversion to the sulfenamide, and reaction with the H,K ATPase. The acid space-dependent accumulation of the PPIs gives them a large margin of safety, and the acid-catalyzed conversion to the cationic SH-reactive sulfenamide produces the long-lasting covalent inhibition of the acid pump. The R moieties represent different substituents at these positions. |
The pyridine of timoprazole, the core structure, has a pKa value of 2.9. Although this would seem appropriate for acid-space targeting, this lead compound was unstable at neutral pH due to the higher nucleophilic reactivity of the pyridine N, an undesirable property when considering either biologic targeting or formulation stability, because the sulfenamide would form spontaneously at neutral pH.
The compounds undergo an acid-catalyzed conversion to a relatively acidstable sulfenamide. This is probably the active compound in free solution, because methylation of the benzimidazole N inactivates the drugs. The molecular rearrangement depends on the presence of a deprotonated pyridine N and a protonated benzimidazole N. The reaction involves a nucleophilic attack by the unprotonated pyridine N on the 2C of the benzimidazole; therefore, an increase of the electrophilicity of this 2C is also required. This is achieved by protonation of the benzimidazole N. The figure illustrates the general chemical mechanism of all current PPIs. The property of acid activation of the PPIs that occurs rapidly at pH of less than 4.0 adds to the safety margin of these drugs.
An alkoxy group at the pyridine 4 (green R”) position flanked by a substituent at the 3 position is the substitution pattern common to all the PPI drugs giving the best combination of efficacy and selectivity. A decrease in the nucleophilicity of the pyridine N (a lower pKa) was able to increase neutral pH stability. In the case of omeprazole, this was achieved by the addition of a methyl group at the 5 position; in lansoprazole, the fluorination of the ethoxy moiety also contributes to this decreased nucleophilicity, and in pantoprazole this was performed by the addition of a second methoxy substitution in the 3
position. This substitution also increases the polarity in this region of the molecule, which may play a role in the cysteine selectivity of this particular drug (see below). The substitution in rabeprazole results in a pK of 4.9, which may contribute to its neutral pH instability but will increase the accumulation in the parietal cell canaliculus.
position. This substitution also increases the polarity in this region of the molecule, which may play a role in the cysteine selectivity of this particular drug (see below). The substitution in rabeprazole results in a pK of 4.9, which may contribute to its neutral pH instability but will increase the accumulation in the parietal cell canaliculus.
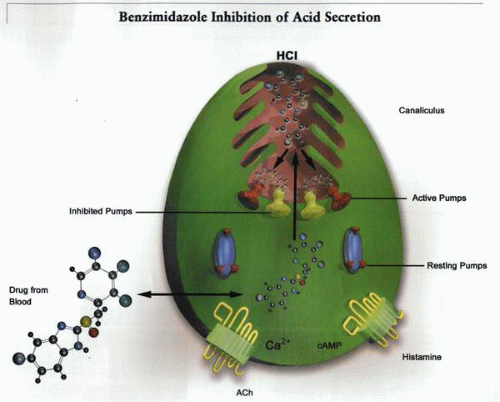 This illustrates the general mechanism for benzimidazole inhibition of gastric acid secretion. The PPI is absorbed from the duodenum and passes via the blood into the parietal cell. If the cell is secreting acid, the prodrug accumulates in the canaliculus and undergoes acid-catalyzed conversion to the sulfenamide, whereon it reacts with cysteines in the active H,K ATPase molecules. |
The rate-limiting step in the formation of the sulfenamide in solution is the formation of the spiro intermediate, which requires folding of the prodrug to allow approach of the pyridine N to the benzimidazole N with perhaps intramolecular proton transfer. This is followed by an attack of the pyridine N on the 2C of the benzimidazole. Access to the transition state is also important. For example, the 3 CH3 substitution favors a folding and interaction of the two ring systems, whereas a 6 CH3 substitution appears to hinder folding and, hence, activation. This latter reactivity can be modified by substitution in the benzimidazole moiety, but this is less important than substitution in the pyridine ring. The difluoromethoxy substitution used in pantoprazole reduces the pKa of the benzimidazole N, explaining the greater neutral pH stability of this compound relative to the other PPIs.
The pH activation curve for the four compounds differs significantly. They are all rapidly converted to the sulfenamide below a pH of 2.0. However, the half-maximal rate of activation is found at a pH of 3.0 for pantoprazole, a pH
of 4.0 for omeprazole and lansoprazole, and a pH of 5.0 for rabeprazole. Protein binding of these drugs may markedly alter the rate of conversion to the sulfenamide, resulting in an unexpected efficacy of pH-unstable compounds.
of 4.0 for omeprazole and lansoprazole, and a pH of 5.0 for rabeprazole. Protein binding of these drugs may markedly alter the rate of conversion to the sulfenamide, resulting in an unexpected efficacy of pH-unstable compounds.
The sulfenamide that is formed from all of these drugs is a permanent cation. This is of benefit because the compound retains its membrane impermeability even after acid secretion into the canaliculus has been inhibited. It is unlikely, therefore, that the active compound can get to the parietal cell cytosol in vivo, adding to the safety profile of these compounds.
This property also ensures targeting only to those SH groups accessible from the luminal or extracytoplasmic surface of the gastric acid pump. The sulfenamide is stable in the absence of SH groups and in acidic medium and is very unstable at neutral pH. This property allowed isolation of the omeprazole sulfenamide in solution, and it also adds to the safety profile of the drugs in that, were the sulfenamide to enter the cytoplasm of a cell, it would have a half-life in the millisecond range.
The reaction of the sulfenamide with SH groups in solution is diffusion limited. However, this does not necessarily extend to the situation in vivo, where the compact structure of the membrane and extracytosolic domains of the H,K ATPase have to be taken into account. This three-dimensional structure of the ATPase could, in principle, dictate derivatization of cysteines that are easily reached from acidic solution and those that are not.
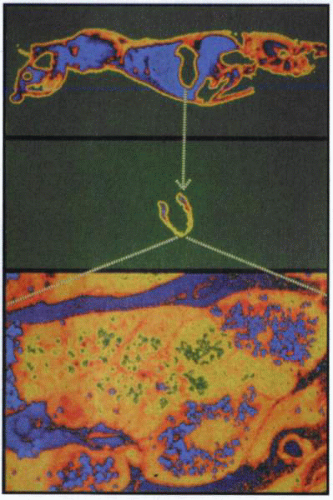 Whole-body autoradiography of a mouse after administration of labeled omeprazole, showing initially a general distribution, but then, at a later time, labeling is confined to the stomach and therein to the parietal cell, demonstrating the remarkable specificity of PPIs. |
Biologic mechanism of PPIs
1. Cellular
The chemistry of the PPIs dictates reaction with electrophilic groups in amino acids after acid-catalyzed conversion of the prodrug. The reactive group is the free −SH of cysteine, which can then form a disulfide bond with the sulfenic acid or sulfenamide derivative of the PPI. In principle, cysteine disulfides can also react, but reformation of the native disulfide would eliminate the bound drug.
There are five potentially reactive cysteines in the catalytic subunit and three disulfide bonds in the beta subunit. After development of omeprazole and knowledge of its chemistry, it was important to show that the mechanism deduced from its chemistry applied to the biologic situation.
Whole-body autoradiography of a mouse injected intravenously with tritiated omeprazole showed general labeling after 5 minutes, but after some hours, labeling was found only in the stomach. Higher magnification showed that labeling was present only in the parietal cell. This means that not only was there highly targeted accumulation of the drug, but also that the binding in the parietal cell was covalent, because it remained for several hours.
Reaction of omeprazole with isolated gastric glands results in inhibition of acid secretion. Treatment with thiol-reducing agents reversed the inhibition in the parietal cell. Omeprazole therefore depends on formation of disulfide bonds for its inhibitory action on gastric acid secretion, as deduced from its chemistry.
Both cysteine and glutathione were effective, and glutathione depletion increased the efficacy of omeprazole.
Both cysteine and glutathione were effective, and glutathione depletion increased the efficacy of omeprazole.
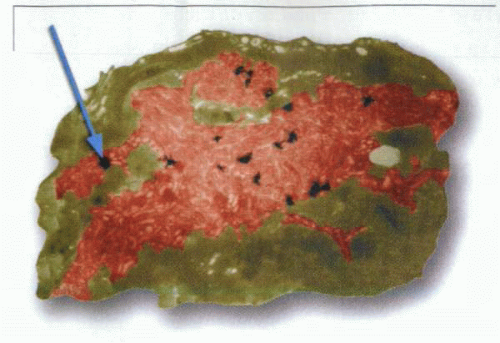 A high-resolution micrograph of a stimulated parietal cell after inhibition by radioactive omeprazole, showing the localization of the binding of this PPI exclusively to the active secretory canaliculus. The dark grains indicate the covalent binding of acid-activated omeprazole to the pump in the secretory canaliculus. |
Treatment of isolated gastric glands with tritiated omeprazole under nonsecreting and acid-secreting conditions allowed analysis of the distribution of the drug as a function of time. The drug showed labeling mainly under acid-secreting conditions and initially labeled only the canalicular compartment, as illustrated in the figure.
In the unstimulated state, there is no labeling of the canaliculus until approximately 30 minutes after introduction of omeprazole. This is due to a basal level of acid secretion.
With stimulation, there is rapid labeling of the canaliculus, with a reaction half-time (t1/2) of approximately 10 minutes. The tubules are not labeled until approximately 30 minutes. Hence, omeprazole reacts only with the pump in the membrane of the acidic canaliculus. Labeling of the cytoplasmic tubules depends on retrieval of the pump from the canalicular membrane.
The mechanism of inhibition of the gastric H,K ATPase in vivo that results from these experimental data suggests that the drug, provided it is given in a gastric acid-protected form, reaches the cytoplasm of the cell via the circulation. It then is accumulated in the canaliculus only if the cell is secreting and then reacts only with the active pumps present in the membrane lining the canaliculus, as illustrated.
2. Enzymic
Treatment of a rabbit with radioactive omeprazole, isolation of gastric membranes, and separation using SDS PAGE, showed that only the catalytic subunit of the H,K ATPase was labeled. Hence, the drug is specific for the alpha subunit of the pump. The general mechanism of pump inhibition by these compounds is illustrated. The drug is accumulated on the luminal face of the pump, where it undergoes acid conversion to the sulfenamide, which then reacts with one or more exposed cysteines on the luminal surface of the enzyme.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


