Infection and Its Consequences
Gastric acid secretion after infection
Acute phase
In early infection, acid secretion decreases, and at least two acid-inhibitory substances have been purified from H. pylori, with one partially sequenced. It appears to represent a metabolic gene. There is inevitably an acute gastritis observed in animal models, with ingress of a variety of inflammatory cells into the submucosa. The reason for the gastritis is obscure, but it is found after infection by any strain of H. pylori. It seems possible that NH3 production is the explanation, because all strains that are infective produce large quantities of NH3 that would tend to alkalinize the interior of gastric epithelial cells.
The addition and removal of NH4Cl has large effects on cell pH. With addition, NH3 is rapidly permeable across cell membranes relative to NH4+ and, on entering a cell, is protonated to NH4+, hence alkalinizing the cell. When the salt is removed, NH3 leaves the cell, and acidification results. Alkalinization of the cells increases intracellular Ca2+, which in turn could result in the production and secretion of cytokines such as IL-8 and others, resulting in the inflammation. From the urease properties defined above, NH3 production would be much larger and constant between pH 6.2 and 2.5. However, at acidic pH, the NH3/NH4+ ratio would decline, resulting in lower concentrations of the permeable NH3. But if there were periods of elevated pH or regions where gastric acid containing NH3 were rapidly neutralized, then the concentration of NH3 would rise, permeate cells, and elevate internal pH and, thus, intracellular calcium. This hypothesis suggests that NH3 production in parallel with varying acidity could account for many of the sequelae of infection.
Chronic phase
It has been known for many years that duodenal ulcer patients have, on the average, higher acid output than do patients without duodenal ulcer. Gastric ulcer patients tend to have lower than normal acidity. Counting the number of parietal cells has shown that duodenal ulcer patients have more parietal cells, thus explaining the acid-output data.
The gastric effects of H. pylori depend in part on the site of infection, be it antral or fundic. Infection, therefore, results in either antral or fundic gastritis. In the case of antral infection, there is a decrease of somatostatin or the D-cell population, accompanied by hypergastrinemia. The latter is thought to be trophic for parietal cells, thus accounting for the higher acid output in duodenal ulcer patients. In the case of infection of the gastric corpus, there is often gastric atrophy with loss of acid-secreting cells. This would result in a reduced level of acid secretion in those patients with gastric atrophy.
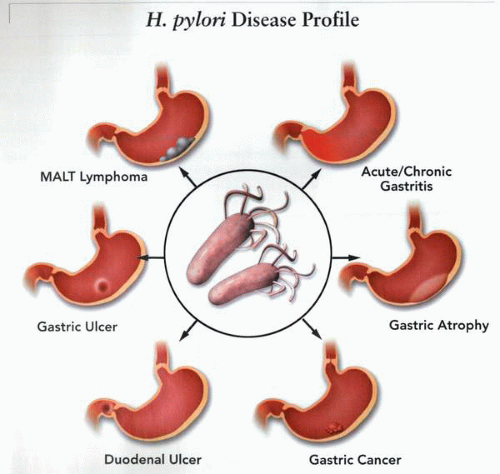 The different potential clinical and pathologic manifestations of infection with H. pylori in the gastroduodenal area. Mucosal infection with the organism results in a number of different events, including active chronic inflammation, lymphoid follicle reaction, development of atrophy, and intestinal metaplasia. In addition, it appears that the organism is also the common denominator in the development of duodenal ulcer, gastric ulcer, MALT gastric lymphoma, and, in some circumstances, gastric adenocarcinoma. The latter diverse pathologic events are multifactorial disease processes, and their evolution to some extent is dependent upon the host-response to infection. Thus, host versus H. pylori interactions and the effects on gastric physiologic functions (acid secretion, mucosal integrity, inflammatory response) and exogenous diverse environmental factors together determine the final expression of the disease in a particular individual. |
Pathogenic strains
As discussed above, all strains result in gastritis at the site of colonization, but only approximately 20% of infected individuals acquire peptic ulcer disease. This may be due to variation in the organism or in the host or due to other factors.
The finding that eradication appears to increase acid reflux that, in turn, may associate with Barrett’s esophagus suggests to some that there may be a benefit to gastric infection. It is true that some frequent human genetic abnormalities may confer protection, such as sickle cell hemoglobin-A mutation providing antimalarial properties or cystic fibrosis transport regulator against typhoid. It does appear unlikely that gastric inflammation is of real benefit to the host.
Helicobacter pylori and ulcer site
The duodenal ulceration site is in the first part of the duodenum, usually at a site where it might be supposed that the highest acidity is found. Even now, it
has not been established that H. pylori is actually present at the site of ulceration or whether gastric metaplasia and ectopic parietal cells are associated with the presence of H. pylori at that locale.
has not been established that H. pylori is actually present at the site of ulceration or whether gastric metaplasia and ectopic parietal cells are associated with the presence of H. pylori at that locale.
The site of gastric ulceration is usually in the transition zone between fundus and antrum. In this region, there are still parietal cells and chief cells that may mainly secrete type II pepsinogen. The organism is clearly found in association with gastric ulcers, and it may be that this region of the stomach is more susceptible to the consequences of infection than the more hardy fundus and the non-acid-secreting antrum.
The presence of acid is as important as that of the organism in the generation of peptic ulcers, and given that the apical membranes of gastric cells are relatively acid impermeable, it is likely that the first site of damage is the tight junction between the epithelial cells. This effect may well be due to inflammation. Once the tight junction is damaged, allowing acid back diffusion, further damage can result in the back diffusion of pepsin.
The combination of infection, inflammation, and acid and pepsin back diffusion results in ulcer development.
Helicobacter pylori and its gastric environment
The organism is found at the gastric surface and within the gastric mucus. There is a degree of controversy about the most frequent site of habitation and as to what the conditions are at the site of habitation. Many believe that the gastric surface is close to neutrality, thus allowing colonization. However, direct measurement has shown that at a luminal pH of 2.0 or less, no pH gradient can be detected at the gastric surface. Because mean diurnal pH is 1.4 in normal people, it is likely that at times during the day, the environment of the organism is genuinely acidic, and its acid-adaptive mechanisms must come into play. Further, because UreI-deletion mutants can inhabit the stomach when acid secretion is inhibited but disappear when acid secretion is allowed to return, clearly the pH of the environment must be less than 4.0 for some considerable time.
It is not known where the first site of colonization is found after infection. It seems that infection often occurs in childhood, perhaps before acid secretion reaches adult levels, and, therefore, either antral or fundic infection could occur. It also seems that infection per se transiently reduces acid secretion by the human stomach, thereby enabling better colonization.
The organisms are found with mainly antral colonization when antral gastritis arises (the more frequent manifestation) or with fundic colonization (when fundic gastritis arises, perhaps a later stage of the disease). They are also often associated with the tight junction regions, perhaps because of the higher urea concentration. Organisms are attached to cells by a pedicle with morphologic alterations at the cellular side of the pedicle. Whether there are specialized outer-membrane proteins then associated with the pedicle, enabling more direct entry of NH3, or perhaps even proteins by type III secretion, is not known.
Several papers have claimed that treatment with PPIs reduces the number of organisms in the antrum and increases their level in the fundus. Inhibition
of acid secretion to the level expected from PPI treatment in principle could reduce acidity on the antral surface to a level at which urease activity could be toxic to the organism. An increase of pH on the fundic surface, improving the habitat there for the bacteria, could increase bacterial frequency in this region of the stomach. Because there is variation in the response to PPIs, there may also be variation in the effect of PPIs on localization of H. pylori in the stomach.
of acid secretion to the level expected from PPI treatment in principle could reduce acidity on the antral surface to a level at which urease activity could be toxic to the organism. An increase of pH on the fundic surface, improving the habitat there for the bacteria, could increase bacterial frequency in this region of the stomach. Because there is variation in the response to PPIs, there may also be variation in the effect of PPIs on localization of H. pylori in the stomach.
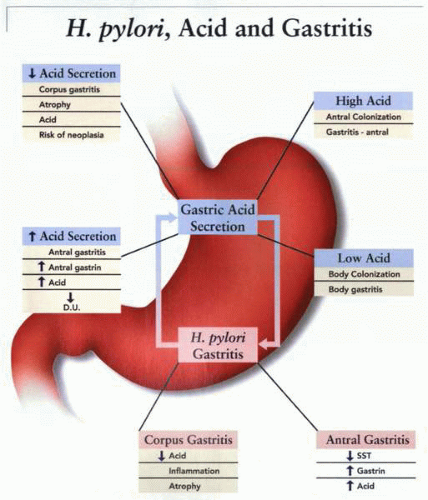 The nature of alterations in acid secretion and different stages of mucosal inflammation that may occur consequent upon H. pylori infection. In patients with a high gastric acid output, colonization may be limited to the antrum, whereas patients with a low gastric acid output are characterized by a body (corpus)-predominant gastritis. The former is associated with a downregulation of somatostatin (SST) secretion, a concomitant increase in gastrin, and a continued high acid secretory response, which may ultimately evolve into a duodenal ulcer phenotype in some patients. Infection of the corpus and a low acid secretory response is associated with mucosal alterations such as atrophy and an increased risk of neoplasia. |
Most agree that fundic infection is able to lead to atrophy, metaplasia, and perhaps cancer. If, indeed, PPI treatment results in relocation of infection to the fundus, there is a strong case to be made for eradication in those patients undergoing chronic therapy with PPIs for GERD.
Serial biopsies taken over many years from patients infected with H. pylori indicate that the long-term consequences of infection may include gastric atrophy and intestinal metaplasia, and, by implication from earlier studies, these may lead to dysplasia and gastric cancer. Although controversial, the concept that decreased acid secretion and gastric atrophy are intimately related is an old one. Although it was previously held that acid inhibition was the result and not the cause of atrophic gastritis, recent data have suggested that if infection
with H. pylori is present, acid inhibition may result in accelerated atrophy. The mechanism of this effect remains obscure, but pharmacologic and surgical reductions of gastric acid secretion are both associated with a more severe inflammatory response to H. pylori, which may lead to more severe epithelial cell damage. Interpretation of the data remains difficult, in part because of the problems in defining gastric atrophy and in part because of age mismatch in the cohorts studied.
with H. pylori is present, acid inhibition may result in accelerated atrophy. The mechanism of this effect remains obscure, but pharmacologic and surgical reductions of gastric acid secretion are both associated with a more severe inflammatory response to H. pylori, which may lead to more severe epithelial cell damage. Interpretation of the data remains difficult, in part because of the problems in defining gastric atrophy and in part because of age mismatch in the cohorts studied.
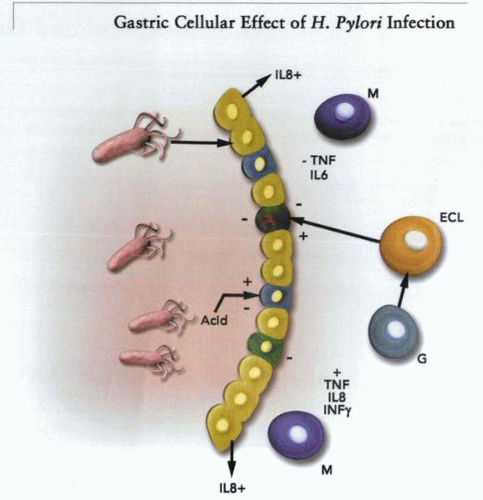 The organism affects a number of cell types in both the antrum and the fundus. These include at least the neuroendocrine G, D, and ECL cells and the immune system. Stimulation of G-cell function (increase in gastrin release) activates the ECL cell to produce histamine that drives parietal cell acid secretion. This effect may be further modulated by the cascade of cytokines that are released by epithelial cells damaged by the organism or its products. Of the neuroendocrine cell acid regulators, ECL cell function is impaired by interleukins. In addition, the gastric T-cell immune response is directed towards the Th1 pathway, which is associated with an enhanced cell-mediated immunity that results in damage (atrophy) to the gastric epithelium. |
The recent publication of the updated Sydney system for the classification and grading of gastritis may be helpful in future studies of atrophy. However, whether this scheme will be more clinically useful than its predecessors remains to be evaluated. Nevertheless, the attempt to objectively define gastritis using visual analog scales is a significant advance and may add considerably to the objectivity of the assessment. However, there is still considerable debate concerning the reversibility of atrophy, whether functional or morphologic; most would argue that atrophy is not reversible. Unfortunately, due to potential sampling errors in follow-up biopsy studies, convincing data are still lacking, and, therefore, debate on this issue will still continue.
Many studies have established that infection with H. pylori and the secondary mucosal inflammatory response increase gastric epithelial cell proliferation.
This may be a necessary step in the process of gastric carcinogenesis, as for many other malignancies. H. pylori probably does not increase proliferation directly; increased cell proliferation is more likely a response to apoptosis (programmed cell death) induced by the organism or the inflammatory response. After a compensatory hyperproliferative response, the balance between apoptosis and proliferation may determine whether ulcers and atrophy develop or, conversely, whether mucosal mass grows in an unrestrained fashion. Some information is available from animal models. For example, H. felis infection in mice increases cell proliferation, particularly of mucous neck cells, but decreases the number of parietal cells. The hyperproliferative response is more extreme in animals that are homozygous for p53, suggesting that H. pylori may act in concert with other oncogenes and tumor-suppressor genes to produce neoplasia. Although little is known about the effect of H. pylori on the normal gastric cell cycle, it has been demonstrated that the lipopolysaccharide of Helicobacter displays synergism in gastrin-mediated increased DNA synthesis in ECL cells.
This may be a necessary step in the process of gastric carcinogenesis, as for many other malignancies. H. pylori probably does not increase proliferation directly; increased cell proliferation is more likely a response to apoptosis (programmed cell death) induced by the organism or the inflammatory response. After a compensatory hyperproliferative response, the balance between apoptosis and proliferation may determine whether ulcers and atrophy develop or, conversely, whether mucosal mass grows in an unrestrained fashion. Some information is available from animal models. For example, H. felis infection in mice increases cell proliferation, particularly of mucous neck cells, but decreases the number of parietal cells. The hyperproliferative response is more extreme in animals that are homozygous for p53, suggesting that H. pylori may act in concert with other oncogenes and tumor-suppressor genes to produce neoplasia. Although little is known about the effect of H. pylori on the normal gastric cell cycle, it has been demonstrated that the lipopolysaccharide of Helicobacter displays synergism in gastrin-mediated increased DNA synthesis in ECL cells.
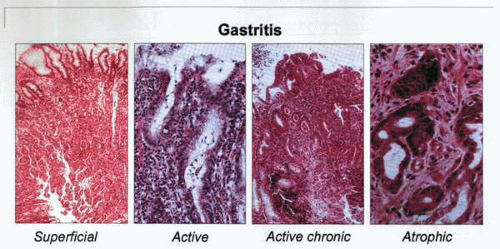 The different histologic types of gastritis identifiable during various stages of H. pylori infection of the gastric mucosa. Superficial gastritis is followed in the majority of patients with active and chronic active gastritis. This is followed, over time and only in a subset of patients, with the development of intestinal metaplasia and atrophy. It has been established that both environmental and host-related factors play significant roles in the evolution of the patient’s gastritis “profile.” |
Because the ECL cell is a crucial link between gastrin and acid in the normal stomach, the interaction of H. pylori with this cell may throw light on some of the discrepant information regarding the effects of H. pylori on gastrin and acid secretion. Whether the ECL cells are exposed to H. pylori lipopolysaccharide directly is unclear, because the ECL cells are not thought to be in communication with the gastric lumen. Nevertheless, lipopolysaccharide is present in measurable quantities in the blood stream and may impinge on the ECL cell in the same fashion that gastrin does. It is possible that mucosal damage induced by H. pylori and disruption of tight junctions may also facilitate access. Either way, it may be that the population of ECL cells does increase in H. pylori infection, and, in combination with PPIs especially, micronodular carcinoids may develop.
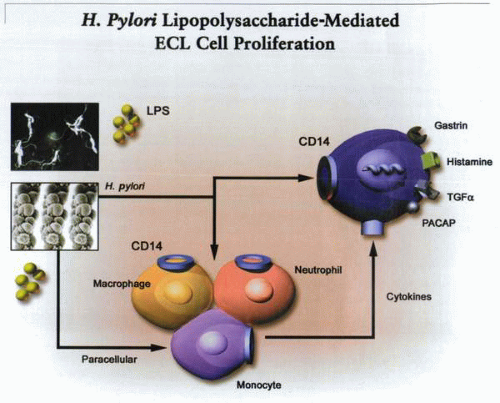 A cartoon of the possible mechanisms by which H. pylori lipopolysaccharide may result in stimulation of ECL cell proliferation and secretion. |
Acid activation of urease in the stomach will result in the production of NH3, which will rapidly convert to NH4+. At pH 4.0, for example, with a pKa of 9.0, there will be a 105-fold excess of NH4+. However, if acid secretion slows or a 10-mM NH4Cl solution is emptied into the duodenum at pH 7.0, there will be a 1,000-fold increase in NH3 concentration. This would be sufficient to alkalinize the cell and elevate intracellular calcium, perhaps to levels at which cytotoxic effects could be observed, as shown in the following figure.
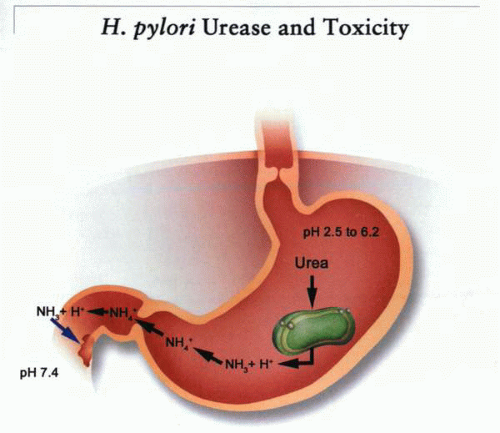 A conceptual model of the means whereby ammonia production from urease activated by gastric acidity may damage the mucosa. In acid, the impermeant ammonium ion is generated, but then with neutralization, the concentration of the diffusible ammonia increases. Entry of this into cells will result in alkalinization and perhaps cytokine release. |
Mucosal-associated lymphoid tissue (MALT) lymphoma
H. pylori vaulted into a position of considerable clinical relevance with the recognition of its critical association with gastroduodenal ulceration. The further observation that the resolution of the genesis of mucosal ulceration might lie with its eradication almost paled in significance when it became evident that the organism was the probable cause of a form of gastric neoplasm. Indeed, the link between H. pylori infection and gastric MALT lymphoma is similar to that between the putative small intestinal infection and IPSID (immuno proliferative small intestinal disease). Because the normal stomach possesses no mucosa-associated lymphoid tissue, the origin of gastric lymphoma is enigmatic. Nevertheless, it is apparent that as a sequel of H. pylori infection, MALT, comprised of lymphoid follicles and a lymphoepithelium, accumulates in the gastric mucosa. The organism has been identified in gastric lymphoma, and the presence of H. pylori in the stomach is directly associated with MALT lymphoma. Furthermore, in areas where there is a high prevalence of H. pylori infection, the likelihood that individuals will develop gastric lymphoma is significantly increased. Because the stomach normally possesses no lymphoid tissue in the gastric mucosa, the accumulation of MALT is almost pathognomonic of H. pylori infection. The proposed role of H. pylori presented antigens in driving the lymphoid tissue hyperplasia, and its transformation into a neoplasm is consistent with current clinical observations. Thus, early antibiotic therapy has been associated with eradication not only of the H. pylori but also of the lymphomatous disease. Alternatively, if the lymphoma has reached an H. pylori-independent phase or has transformed from
its low-grade status to a high-grade MALT lymphoma, eradication therapy is not effective.
its low-grade status to a high-grade MALT lymphoma, eradication therapy is not effective.
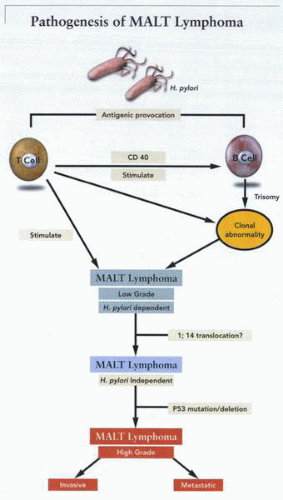 A schematic outline of the pathogenesis of MALT lymphoma due to stimulation of B- or T-cell proliferation by H. pylori antigens, followed by neoplastic transformation. |
The gastrointestinal tract is the most common site of primary extranodal lymphoma, and gastric lymphoma accounts for the majority of cases. It is of importance to differentiate primary gastric lymphoma from secondary involvement of the stomach by nodal lymphoma, which occurs relatively commonly. The precise incidence of primary gastric lymphoma is difficult to ascertain, given the widely different diagnostic criteria that have been used as well as the considerable geographic variation in incidences. Overall, there is evidence to support the fact that gastric lymphoma may be increasing in incidence, although the incidence of H. pylori is decreasing. It is necessary, however, to define primary gastric lymphoma as lymphoma occurring in the stomach with or without the presence of regional lymph nodes, with the stomach being the site of the majority of the disease, if not the only site.
Hodgkin’s disease rarely occurs in the stomach, and the great majority of primary gastric lymphomas are B-cell tumors. Nevertheless, T-cell lymphomas do occur, although they are extremely rare. For the most part, the histopathologic features of low-grade primary gastric lymphoma—MALT—recapitulate the structure of Peyer’s patches rather than lymph nodes. In certain instances, low-grade MALT lymphomas may transform to high-grade disease, and it is likely that most instances of this entity represent evolution, because the tumors are derived from the same B-cell lineage. What is apparent, however, is that gastric MALT lymphomas do not share any of the features common to nodal lymphomas but instead exhibit a marked increase in the frequency of trisomy 3. Similarly, the gastric MALT lymphomas differ from their nodal counterparts in that their behavior is usually quite favorable.
The low-grade gastric lymphomas, which have been characterized as MALT, usually occur in individuals older than 50 years, with a peak in the seventh decade. Nevertheless, instances of such disease have been described at almost all
ages. There appears to be a slight male predominance (1.5:1.0). More often than not, the symptoms are of a nonspecific nature, with a central dyspeptic component rather than any specific signs, as might occur with a gastric adenocarcinoma. At endoscopy, the findings are usually those of a nonspecific gastritis with erosions or ulceration, although a mass lesion is occasionally identifiable. It is unusual to be able to detect extraabdominal dissemination, although such events have been recorded. In contrast to the low-grade MALT lymphomas, patients presenting with the high-grade B-cell gastric lymphomas usually do so at a slightly older age (64 years versus 55 years). In these patients, the clinical presentation more commonly represents that of a gastric adenocarcinoma with pain, weight loss, and anemia being the most common presentations, although perforation rarely may occur as the initial clinical event. At endoscopy, an obvious tumor mass is usually evident, with ulceration in many instances. For the most part, gastric lymphomas involve the antrum, but they may occur at any site in the stomach. The low-grade MALT lymphomas usually are flat infiltrative lesions, which are often difficult to diagnose and often require multiple or double-level biopsies. The high-grade gastric lymphomas are more commonly large and bulky tumors with considerable infiltration.
ages. There appears to be a slight male predominance (1.5:1.0). More often than not, the symptoms are of a nonspecific nature, with a central dyspeptic component rather than any specific signs, as might occur with a gastric adenocarcinoma. At endoscopy, the findings are usually those of a nonspecific gastritis with erosions or ulceration, although a mass lesion is occasionally identifiable. It is unusual to be able to detect extraabdominal dissemination, although such events have been recorded. In contrast to the low-grade MALT lymphomas, patients presenting with the high-grade B-cell gastric lymphomas usually do so at a slightly older age (64 years versus 55 years). In these patients, the clinical presentation more commonly represents that of a gastric adenocarcinoma with pain, weight loss, and anemia being the most common presentations, although perforation rarely may occur as the initial clinical event. At endoscopy, an obvious tumor mass is usually evident, with ulceration in many instances. For the most part, gastric lymphomas involve the antrum, but they may occur at any site in the stomach. The low-grade MALT lymphomas usually are flat infiltrative lesions, which are often difficult to diagnose and often require multiple or double-level biopsies. The high-grade gastric lymphomas are more commonly large and bulky tumors with considerable infiltration.
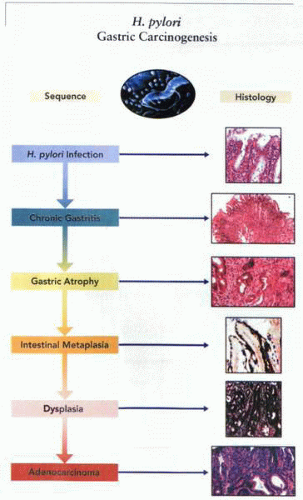 Possible histology of the sequence of events due to infection by H. pylori leading to gastric adenocarcinoma. |
The low-grade MALT lymphomas produce a histologic appearance closely simulating that of a Peyer’s patch. Thus, the lymphoma infiltrates around and between reactive follicles in the region corresponding to the marginal zone of a Peyer’s patch and spreads diffusely into the surrounding mucosa. Tumor cells are usually small to medium in size, with moderately abundant cytoplasm with nuclei that have an irregular outline closely resembling the nuclei of centrocytes. An important histologic feature of low-grade MALT lymphomas is the presence of lymphoepithelial lesions formed by the invasion of individual gastric glands by aggregates of tumor cells. At a later stage, this is associated with disintegration of the glandular epithelium and eosinophilic degeneration. Occasionally areas of a low-grade lymphoma may be replaced by a high-grade lymphoma, suggesting transformation from one form to the other. The presence of confluent clusters or sheets of transformed cells is strongly indicative of transformation to a high-grade lymphoma lesion.
Low-grade MALT lymphomas are seldom disseminated at the time of diagnosis and rarely involve lymph nodes or bone marrow. As a result, prolonged survival is common, with figures reaching as high 91% at 5 years and 75% at 10 years if surgical resection is used. Nodal low-grade B-cell lymphomas are usually widely disseminated at diagnosis, with the majority of patients having
bone marrow involvement. Treatment is usually ineffective, and most patients die within 7 to 10 years, often as a result of high-grade transformation. There is controversy as to whether high-grade MALT lymphomas have as favorable a prognosis. On balance, it appears that the higher the grade of lymphoma, the less favorable the outcome is likely to be.
bone marrow involvement. Treatment is usually ineffective, and most patients die within 7 to 10 years, often as a result of high-grade transformation. There is controversy as to whether high-grade MALT lymphomas have as favorable a prognosis. On balance, it appears that the higher the grade of lymphoma, the less favorable the outcome is likely to be.
Low-grade MALT lymphomas commonly involve the local draining lymph nodes; if peripheral spreading occurs, it is a late event. This results in a favorable prognosis quite unlike that of the indolent yet progressive disease pattern associated with disease-disseminated low-grade B-cell lymphomas of lymph nodes. In fact, low-grade MALT lymphomas tend to remain localized to their site of origin for prolonged periods, although the reason for this is not clearly defined. It has been proposed that this type of lesion may not even be a malignant lymphoma but represents either a hyperplasia or a “pseudolymphoma.” One suggestion is that the proliferation of the MALT represents the presence of a local antigen presented by H. pylori,




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
