Hereditary Paraganglioma/Pheochromocytoma Syndromes
Vania Nosé, MD, PhD
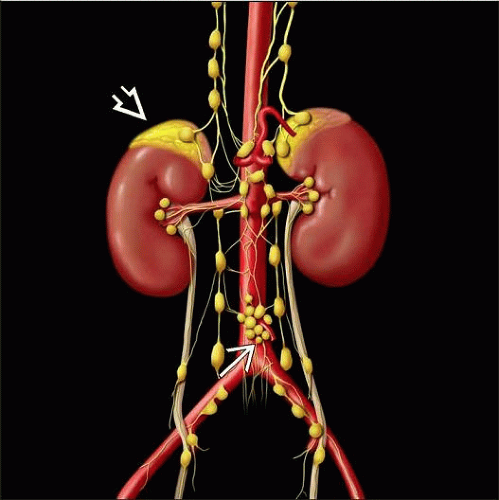 Graphic shows paraganglia and neuroendocrine tissues symmetrically distributed along the paravertebral axis in the abdomen, including the organ of Zuckerkandl  and the adrenal medulla and the adrenal medulla  . . |
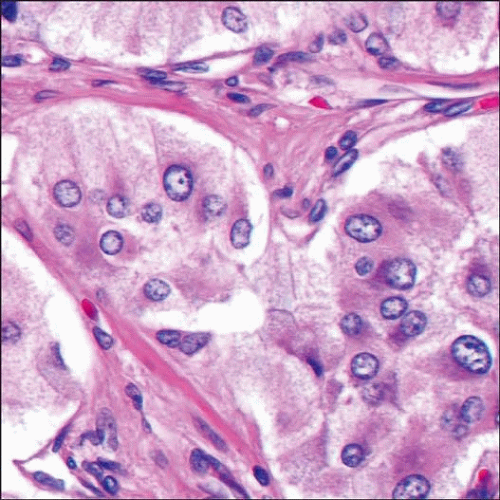 This pheochromocytoma (PCC) has the characteristic alveolar pattern (zellballen) with variably sized nests of tumor cells surrounded by thin-walled vessels and thin bands of fibrous tissue. |
TERMINOLOGY
Abbreviations
Hereditary paraganglioma/pheochromocytoma (PGL/PCC) syndromes
Paraganglioma (PGL)
Pheochromocytoma (PCC)
Definitions
PCCs and PGLs are neuroendocrine tumors that arise in adrenal medulla or extraadrenal sympathetic and parasympathetic paraganglia
Occur sporadically or as part of different hereditary tumor syndromes
Tumors arising within adrenal medulla are known as PCCs; histologically identical tumors arising elsewhere are termed PGLs
Hereditary PGL/PCC syndromes are characterized by presence of PGL &/or PCC that occur as part of a familial syndrome
> 30% of PCCs and PGLs are currently believed to be caused by germline mutations and several novel susceptibility genes have recently been discovered
RET, VHL, NF1, SDHA, SDHB, SDHC, SDHD, SDHAF2, KIF1Bβ, TMEM127, and MAX have been associated with hereditary PCC or PGL
Hereditary PGL/PCC syndromes should be considered in all individuals with PGL or PCC with the following findings
Multiple tumors, including bilateral tumors
Multifocal with multiple synchronous or metachronous tumors
Early onset (age < 40 years)
Family history of such tumors
Familial PGL/PCC syndrome is term restricted to tumors from germline mutations in SDHx genes
Simplex cases: Many individuals with a hereditary PGL/PCC syndrome may present with solitary tumor of head or neck, thorax, abdomen, adrenal, or pelvis and no family history of the disorder
In PGL/PCC that appear to be sporadic based on the absence of a family history, rate of occult germline mutation is said to be ˜ 12% and ranges from 7.5-24%
Syndromes Characterized by Susceptibility to PCC and PGL
Most tumors were known to be associated with multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau disease (VHL), and neurofibromatosis type 1 (NF1)
More recently, mutations in genes encoding different subunits of succinate dehydrogenase (SDH) complex have been linked to familial PGL/PCC syndrome (PGL1, 2, 3, and 4)
Small fraction is associated with other syndromes (e.g., Carney triad, Carney-Stratakis syndrome, MEN1)
Several other genes have recently been added to the list (associated with unknown hereditary PGL/PCC)
Kinesin family member 1B (KIF1B)
EGL-9 homolog 1 (EGLN1), also termed PHD2
Transmembrane protein 127 (TMEM127)
MYC-associated factor X (MAX)
GENETICS
MEN2
Autosomal dominant syndrome caused by mutation of RET proto-oncogene
Activating RET mutation predisposes to PCC, which is often bilateral and recurrent
Low risk of malignancy
MEN2 prevalence is estimated at 1:30,000
MEN2 often suspected on basis of family history; individuals with PCC infrequently present as simplex cases
Clinically, can be divided into 3 types: MEN2A (55% of all cases), MEN2B (5-10%), and familial medullary thyroid carcinoma (FMTC, 35-40%)
MEN2A and MEN2B patients have almost 100% risk of developing medullary thyroid carcinoma
˜ 50% of individuals with MEN2A and MEN2B develop PCC
Subtype FMTC has medullary thyroid carcinoma as its only feature
Familial PGL/PCC Syndromes
Germline mutations in SDHx genes give rise to familial PGL/PCC syndrome, sometimes only referred to as familial PGL
Prevalence of PGL/PCC syndrome is unknown, but a review of ˜ 13% of all PGL/PCC cases gives an estimate of 1:50,000 to 1:20,000; majority represented by PGL1 and PGL4
Associated with germline mutations in genes encoding subunits of SDH enzyme complex in context of familial PGL syndromes; PGL1, PGL2, PGL3, and PGL4 caused by mutations in SDHD, SDHAF2, SDHC, and SDHB genes, respectively
PGL2 is caused by mutations in SDHAF2/SDH5, which encodes for a molecule that is an accessory to the function of the SDH enzyme and its SDHA subunit
Mutations were recently found in SDHA subunit in a limited number of patients with PGL &/or PCC
SDHB mutations in particular may also predispose to thyroid and renal cancer, and possibly other tumors
Patients harboring SDHB mutation are at increased risk of malignancy
Genotype–phenotype correlation
People with SDHB, SDHD, and SDHC mutations can develop PCCs or PGLs anywhere in paraganglia
Genotype-phenotype correlations guiding diagnostic testing and patient care
Germline mutations in SDHB are strongly associated with extraadrenal sympathetic PGL
Chromaffin tumors in people with germline SDHB mutations are 6x more likely to be extraadrenal than chromaffin tumors in general
PGL in people with germline SDHB mutation are more likely to become malignant than sporadic PGL or in those with germline SDHD and SDHC mutations
SDHB mutations also predict shorter survival
Up to 50% of people with malignant extraadrenal PGL have a germline SDHB mutation PGL
People with a germline SDHD mutation are more likely to develop head and neck and abdominal PGL compared with people with a germline SDHB mutation
Germline SDHC mutations appear to be primarily associated with head and neck PGL
von Hippel-Lindau Syndrome (VHL)
Autosomal dominant disorder caused by mutation of VHL
Features include retinal angiomas, central nervous system hemangioblastomas, clear cell renal cell carcinoma, pancreatic endocrine tumors, endolymphatic sac tumors, renal, pancreatic and epididymal cysts, and PCCs
Occurs in ˜ 1/36,000 individuals
˜ 10-26% of VHL patients develop PCC or PGL, but risk varies between families
Frequency of PCC in individuals with VHL is 10-20%
Mean age of onset of PCC in VHL is ˜ 30 years
PCCs occur in only 6-9% of individuals with VHL type 1
Prevalence of PCC rises to 40-59% in individuals with VHL type 2
In type 2C VHL, PCCs are sole manifestation of the syndrome (simplex cases)
VHL mutations predispose to unilateral or bilateral PCCs and, much less frequently, to sympathetic or parasympathetic PGLs
˜ 50% of PCCs are bilateral
PCCs in VHL secrete primarily norepinephrine and normetanephrine
˜ 5% Of VHL-related catecholamine secreting tumors become malignant, most commonly extraadrenal sympathetic PGL
Only 3% displayed malignant tumors
Bilateral PCC was seen in 44% of the patients
Mean age at diagnosis of PGL/PCC is 29 years
PCC or PGL is 1st manifestation of VHL disease in 30-55% of cases
VHL can be distinguished from other hereditary PGL/PCC syndromes clinically
NF1
Autosomal dominant disorder caused by mutation of NF1
Major features of NF1 include neurofibromas, café au lait spots, iris hamartomas, and axillary and inguinal freckling
Gastrointestinal stromal tumors (GISTs) and carcinoid tumors may also occur
PCCs and PGLs are not among most common manifestations of NF1 but occur in 0.1-5.7% of patients
PCCs occur in 20-50% of individuals with NF1 and hypertension
NF1-associated PCCs and PGLs typically have characteristics similar to those of sporadic tumors, with a relatively late mean age of onset and ˜ 10% risk of malignancy
Up to 84% of PCC are unilateral
Extraadrenal sympathetic PGL can occur
95% of patients with NF1 had PCC and 6% had PGL; all PGLs were sympathetic
14% of patients displayed bilateral PCC
9% developed malignant disease
Carney Triad (CT)
Rare multitumoral syndrome of unknown etiology
Some SDH-deficient GISTs are driven by classical SDH mutations, but precise mechanisms of tumorigenesis in those associated with Carney triad remain unknown
Usually occurs in young women
Neoplasms affect stomach, lungs, paraganglionic system, adrenal cortex, and esophagus
Triad: Gastric stromal tumor, PGL, and pulmonary chondroma
PCC, adrenal cortical adenoma, and esophageal leiomyoma are also associated
Multifocal tumors develop in affected organs
Mean age at presentation with PGL/PCC is 28 years
92% present with PGL, including both sympathetic and parasympathetic tumors, and ˜ 16% present with PCC
Multiple PGLs are found in 22% of patients and bilateral PCC in 3%
Metastasis occurs in 11% of patients
Carney-Stratakis Syndrome
Mutations in SDHB, SDHC, and SDHD can give rise to Carney-Stratakis syndrome, characterized by dyad of PGLs and GISTs
100% of patients had PGL and 1 patient also presented with unilateral PCC, with a mean age of 33 years at presentation
PGLs occur in head and neck, thorax, and abdomen
Multiple PGLs, which could be both sympathetic and parasympathetic, were seen in 73% of patients
None of the tumors were malignant
MEN1
Caused by mutations in MEN1 gene
MEN1 gene is a 10-exon gene that encodes 610-amino acid protein, menin
Mutation spectrum
> 1,300 different mutations of MEN1 gene have been characterized
Penetrance of MEN1 is high: 45% by age 30, 82% by age 50, 96% by age 70
Spread over entire coding and intronic sequence
> 60% truncating mutation, 20% missense mutation, 10% frame deletions or insertions, 10% others
Most are inactivating
Function is unknown; may act as regulator of gene transcription, cell proliferation, apoptosis, and genome stability
No cases of PGL and only 7 cases of PCC in MEN1 syndrome have been reported in the literature
Reported tumors were unilateral in all cases and malignant in 1 case
Other Genes Involved in PGL/PCC
Several other genes have recently been added to the list (associated with unknown hereditary PGL/PCC)
Kinesin family member 1B (KIF1B); EGL-9 homolog 1 (EGLN1), also termed PHD2; transmembrane protein 127 (TMEM127); and MYC-associated factor X (MAX)
No specific syndrome has been attributed yet, but patients with germline KIF1Bβ mutations seem to be predisposed to at least PCCs and neuroblastomas
Ganglioneuroma, leiomyosarcoma, and lung adenocarcinoma have also been reported in a family with KIF1Bβ mutations
Only 1 PGL patient, suffering from recurrent PGL and erythrocytosis, has been reported to have a germline mutation in EGLN1
Presentation with sympathetic PGL and a recurrent tumor was diagnosed 3 years later, but no metastases have been reported
So far, no specific syndrome has been described for TMEM127
TMEM127 mutations were identified in 2% of the cases considered sporadic, all of which had PCC
96% of patients have PCC and 39% have bilateral PCC
MAX mutations segregate with disease in families with PCC, but no specific syndrome has been described yet
Usually bilateral tumors, early age of onset, &/or familial antecedents with the disease
Notably, 25% of patients showed metastasis at diagnosis, suggesting that MAX mutations are associated with high risk of malignancy
So far, no studies on PGLs have been reported
CLINICAL IMPLICATIONS AND ANCILLARY TESTS
Immunohistochemistry
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


