KEY POINTS
The life span of platelets ranges from 7 to 10 days. Drugs that interfere with platelet function include aspirin, clopidogrel, prasugrel, dipyridamole, and the glycoprotein IIb/IIIa (GP IIb/IIIa) inhibitors. Approximately 5 to 7 days should pass from the time the drug is stopped until an elective procedure is performed.
The acute coagulopathy of trauma results from a combination of activation of protein C and hyperfibrinolysis. It is distinct from disseminated intravascular coagulation, is present on arrival to the emergency department, and is associated with an increase in mortality.
Newer anticoagulants like dabigatran and rivaroxaban have no readily available method of detection of the degree of anticoagulation and may not be readily reversible.
Therapeutic anticoagulation preoperatively and postoperatively is becoming increasingly more common. The patient’s risk of intraoperative and postoperative bleeding should guide the need for reversal of anticoagulation therapy preoperatively and the timing of its reinstatement postoperatively.
Damage control resuscitation has three basic components: permissive hypotension, minimizing crystalloid-based resuscitation, and the administration of predefined blood products.
The need for massive transfusion should be anticipated, and guidelines should be in place to provide early and increased amounts of red blood cells, plasma, and platelets.
BIOLOGY OF HEMOSTASIS
Hemostasis is a complex process whose function is to limit blood loss from an injured vessel. Four major physiologic events participate in the hemostatic process: vascular constriction, platelet plug formation, fibrin formation, and fibrinolysis. Although each tends to be activated in order, the four processes are interrelated so that there is a continuum and multiple reinforcements. The process is shown schematically in Fig. 4-1.
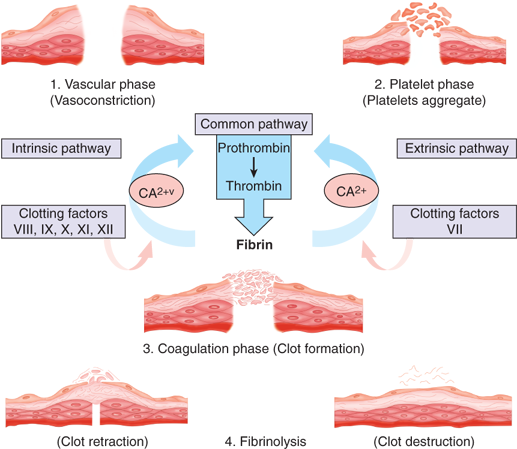
Vascular constriction is the initial response to vessel injury. It is more pronounced in vessels with medial smooth muscles and is dependent on local contraction of smooth muscle. Vasoconstriction is subsequently linked to platelet plug formation. Thromboxane A2 (TXA2) is produced locally at the site if injury via the release of arachidonic acid from platelet membranes and is a potent constrictor of smooth muscle. Similarly, endothelin synthesized by injured endothelium and serotonin (5-hydroxytryptamine [5-HT]) released during platelet aggregation are potent vasoconstrictors. Lastly, bradykinin and fibrinopeptides, which are involved in the coagulation schema, are also capable of contracting vascular smooth muscle.
The extent of vasoconstriction varies with the degree of vessel injury. A small artery with a lateral incision may remain open due to physical forces, whereas a similarly sized vessel that is completely transected may contract to the extent that bleeding ceases spontaneously.
Platelets are anucleate fragments of megakaryocytes. The normal circulating number of platelets ranges between 150,000 and 400,000/μL. Up to 30% of circulating platelets may be sequestered in the spleen. If not consumed in a clotting reaction, platelets are normally removed by the spleen and have an average life span of 7 to 10 days.
Platelets play an integral role in hemostasis by forming a hemostatic plug and by contributing to thrombin formation (Fig. 4-2). Platelets do not normally adhere to each other or to the vessel wall but can form a plug that aids in cessation of bleeding when vascular disruption occurs. Injury to the intimal layer in the vascular wall exposes subendothelial collagen to which platelets adhere. This process requires von Willebrand factor (vWF), a protein in the subendothelium that is lacking in patients with von Willebrand’s disease. vWF binds to glycoprotein (GP) I/IX/V on the platelet membrane. Following adhesion, platelets initiate a release reaction that recruits other platelets from the circulating blood to seal the disrupted vessel. Up to this point, this process is known as primary hemostasis. Platelet aggregation is reversible and is not associated with secretion. Additionally, heparin does not interfere with this reaction, and thus, hemostasis can occur in the heparinized patient. Adenosine diphosphate (ADP) and serotonin are the principal mediators in platelet aggregation.
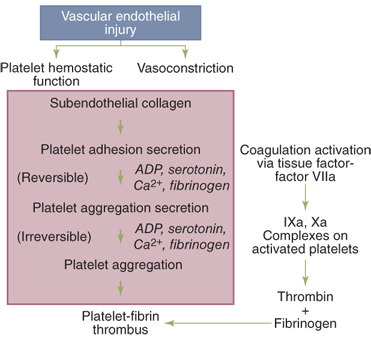
Arachidonic acid released from the platelet membranes is converted by cyclooxygenase to prostaglandin G2 (PGG2) and then to prostaglandin H2 (PGH2), which, in turn, is converted to TXA2. TXA2 has potent vasoconstriction and platelet aggregation effects. Arachidonic acid may also be shuttled to adjacent endothelial cells and converted to prostacyclin (PGI2), which is a vasodilator and acts to inhibit platelet aggregation. Platelet cyclooxygenase is irreversibly inhibited by aspirin and reversibly blocked by nonsteroidal anti-inflammatory agents, but is not affected by cyclooxygenase-2 (COX-2) inhibitors.
In the second wave of platelet aggregation, a release reaction occurs in which several substances including ADP, Ca2+, serotonin, TXA2, and α-granule proteins are discharged. Fibrinogen is a required cofactor for this process, acting as a bridge for the GP IIb/IIIa receptor on the activated platelets. The release reaction results in compaction of the platelets into a plug, a process that is no longer reversible. Thrombospondin, another protein secreted by the α-granule, stabilizes fibrinogen binding to the activated platelet surface and strengthens the platelet-platelet interactions. Platelet factor 4 (PF4) and α-thromboglobulin are also secreted during the release reaction. PF4 is a potent heparin antagonist. The second wave of platelet aggregation is inhibited by aspirin and nonsteroidal anti-inflammatory drugs, by cyclic adenosine monophosphate (cAMP), and by nitric oxide. As a consequence of the release reaction, alterations occur in the phospholipids of the platelet membrane that allow calcium and clotting factors to bind to the platelet surface, forming enzymatically active complexes. The altered lipoprotein surface (sometimes referred to as platelet factor 3) catalyzes reactions that are involved in the conversion of prothrombin (factor II) to thrombin (factor IIa) by activated factor X (Xa) in the presence of factor V and calcium, and it is involved in the reaction by which activated factor IX (IXa), factor VIII, and calcium activate factor X. Platelets may also play a role in the initial activation of factors XI and XII.
Hemostasis involves a complex interplay and combination of interactions between platelets, the endothelium, and multiple circulating or membrane-bound coagulation factors. While a bit simplistic and not reflective of the depth or complexity of these interactions, the coagulation cascade has traditionally been depicted as two possible pathways converging into a single common pathway (Fig. 4-3). While this pathway reflects the basic process and sequences that lead to the formation of a clot, the numerous feedback loops, endothelial interplay, and platelet functions are not included. The intrinsic pathway begins with the activation of factor XII that subsequently activates factors XI, IX, and VIII. In this pathway, each of the primary factors is “intrinsic” to the circulating plasma, whereby no surface is required to initiate the process. In the extrinsic pathway, tissue factor (TF) is released or exposed on the surface of the endothelium, binding to circulating factor VII, facilitating its activation to VIIa. Each of these pathways continues on to a common sequence that begins with the activation of factor X to Xa (in the presence of VIIIa). Subsequently, Xa (with the help of factor Va) converts factor II (prothrombin) to thrombin and then factor I (fibrinogen) to fibrin. Clot formation occurs after fibrin monomers are cross-linked to polymers with the assistance of factor XIII.
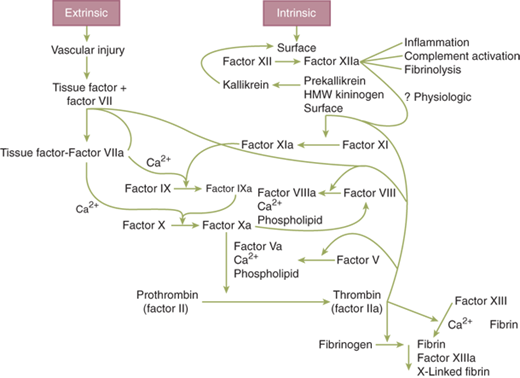
One convenient feature of depicting the coagulation cascade with two merging arms is that commonly used laboratory tests segregate abnormalities of clotting to one of the two arms. An elevated activated partial thromboplastin time (aPTT) is associated with abnormal function of the intrinsic arm of the cascade (II, IX, X, XI, XII), while the prothrombin time (PT) is associated with the extrinsic arm (II, VII, X). Vitamin K deficiency or warfarin use affects factors II, VII, IX, and X
Expanding from the basic concept of Fig. 4-3, the primary pathway for coagulation is initiated by TF exposure following subendothelial injury. Clot propagation ensues with what is a sequence of four similar enzymatic reactions, each involving a proteolytic enzyme generating the next enzyme by cleaving its proenzyme, a phospholipid surface (e.g., platelet membrane) in the presence of ionized calcium, and a helper protein. TF binds to VIIa, and this complex catalyzes the activation of factor X to Xa. This complex is four orders of magnitude more active at converting factor X than is factor VIIa alone and also activates factor IX to IXa. Factor Xa, together with Va, calcium, and phospholipid, composes the prothrombinase complex that converts prothrombin to thrombin. The prothrombinase complex is significantly more effective at catalyzing its substrate than is factor Xa alone. Thrombin is then involved with the conversion of fibrinogen to fibrin and activation of factors V, VII, VIII, XI, and XIII.
In building on the redundancy inherent in the coagulation system, factor VIIIa combines with IXa to form the intrinsic factor complex. Factor IXa is responsible for the bulk of the conversion of factor X to Xa. This complex (VIIIa-IXa) is 50 times more effective at catalyzing factor X activation than is the extrinsic (TF-VIIa) complex and five to six orders of magnitude more effective than factor IXa alone.
Once formed, thrombin leaves the membrane surface and converts fibrinogen by two cleavage steps into fibrin and two small peptides termed fibrinopeptides A and B. Removal of fibrinopeptide A permits end-to-end polymerization of the fibrin molecules, whereas cleavage of fibrinopeptide B allows side-to-side polymerization of the fibrin clot. This latter step is facilitated by thrombin-activatable fibrinolysis inhibitor (TAFI), which acts to stabilize the resultant clot.
In seeking to balance profound bleeding with overwhelming clot burden, several related processes exist to prevent propagation of the clot beyond the site of injury.1 First, feedback inhibition on the coagulation cascade deactivates the enzyme complexes leading to thrombin formation. Thrombomodulin (TM) presented by the endothelium serves as a “thrombin sink” by forming a complex with thrombin, rendering it no longer available to cleave fibrinogen. This then activates protein C (APC) and reduces further thrombin generation by inhibiting factors V and VIII. Second, tissue plasminogen activator (tPA) is released from the endothelium following injury, cleaving plasminogen to initiate fibrinolysis. APC then consumes plasminogen activator inhibitor-1 (PAI-1), leading to increased tPA activity and fibrinolysis. Building on the anticoagulant response to inhibit thrombin formation, tissue factor pathway inhibitor (TFPI) is released, blocking the TF-VIIa complex and reducing the production of factors Xa and IXa. Antithrombin III (AT-III) then neutralizes all of the procoagulant serine proteases and also inhibits the TF-VIIa complex. The most potent mechanism of thrombin inhibition involves the APC system. APC forms a complex with its cofactor, protein S, on a phospholipid surface. This complex then cleaves factors Va and VIIIa so they are no longer able to participate in the formation of TF-VIIa or prothrombinase complexes. This is of interest clinically in the form of a genetic mutation, called factor V Leiden. In this setting, factor V is resistant to cleavage by APC, thereby remaining active as a procoagulant. Patients with factor V Leiden are predisposed to venous thromboembolic events.
Degradation of fibrin clot is accomplished by plasmin, a serine protease derived from the proenzyme plasminogen. Plasmin formation occurs as a result of one of several plasminogen activators. tPA is made by the endothelium and other cells of the vascular wall and is the main circulating form of this family of enzymes. tPA is selective for fibrin-bound plasminogen so that endogenous fibrinolytic activity occurs predominately at the site of clot formation. The other major plasminogen activator, urokinase plasminogen activator (uPA), also produced by endothelial cells as well as by urothelium, is not selective for fibrin-bound plasminogen. Of note, the thrombin-TM complex activates TAFI, leading to a mixed effect on clot stability. In addition to inhibiting fibrinolysis directly, removal of the terminal lysine on the fibrin molecule by TAFI renders the clot more susceptible to lysis by plasmin.
Fibrin clot breakdown (lysis) allows restoration of blood flow during the healing process following injury and begins at the same time clot formation is initiated. Fibrin polymers are degraded by plasmin, a serine protease derived from the proenzyme plasminogen. Plasminogen is converted to plasmin by one of several plasminogen activators, including tPA. Plasmin then degrades the fibrin mesh at various places, leading to the production of circulating fragments, termed fibrin degradation products (FDPs), cleared by other proteases or by the kidney and liver (Fig. 4-4). Fibrinolysis is directed by circulating kinases, tissue activators, and kallikrein present in vascular endothelium. tPA is synthesized by endothelial cells and released by the cells on thrombin stimulation. Bradykinin, a potent endothelial-dependent vasodilator, is cleaved from high molecular weight kininogen by kallikrein and enhances the release of tPA. Both tPA and plasminogen bind to fibrin as it forms, and this trimolecular complex cleaves fibrin very efficiently. After plasmin is generated, however, it cleaves fibrin somewhat less efficiently.
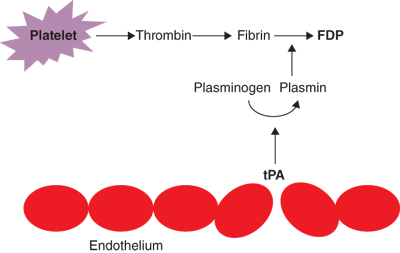
As with clot formation, fibrinolysis is also kept in check through several robust mechanisms. tPA activates plasminogen more efficiently when it is bound to fibrin, so that plasmin is formed selectively on the clot. Plasmin is inhibited by α2-antiplasmin, a protein that is cross-linked to fibrin by factor XIII, which helps to ensure that clot lysis does not occur too quickly. Any circulating plasmin is also inhibited by α2-antiplasmin and circulating tPA or urokinase. Clot lysis yields FDPs including E-nodules and D-dimers. These smaller fragments interfere with normal platelet aggregation, and the larger fragments may be incorporated into the clot in lieu of normal fibrin monomers. This may result in an unstable clot as seen in cases of severe coagulopathy such as hyperfibrinolysis associated with trauma-induced coagulopathy or disseminated intravascular coagulopathy. The presence of D-dimers in the circulation may serve as a marker of thrombosis or other conditions in which a significant activation of the fibrinolytic system is present. Another inhibitor of the fibrinolytic system is TAFI, which removes lysine residues from fibrin that are essential for binding plasminogen.
CONGENITAL FACTOR DEFICIENCIES
Inherited deficiencies of all of the coagulation factors are seen. However, the three most frequent are factor VIII deficiency (hemophilia A and von Willebrand’s disease), factor IX deficiency (hemophilia B or Christmas disease), and factor XI deficiency. Hemophilia A and hemophilia B are inherited as sex-linked recessive disorders with males being affected almost exclusively. The clinical severity of hemophilia A and hemophilia B depends on the measurable level of factor VIII or factor IX in the patient’s plasma. Plasma factor levels less than 1% of normal are considered severe disease, factor levels between 1% and 5% moderately severe disease, and levels between 5% and 30% mild disease. Patients with severe hemophilia have spontaneous bleeds, frequently into joints, leading to crippling arthropathies. Intracranial bleeding, intramuscular hematomas, retroperitoneal hematomas, and gastrointestinal, genitourinary, and retropharyngeal bleeding are added clinical sequelae seen with severe disease. Patients with moderately severe hemophilia have less spontaneous bleeding but are likely to bleed severely after trauma or surgery. Mild hemophiliacs do not bleed spontaneously and have only minor bleeding after major trauma or surgery. Since platelet function is normal in hemophiliacs, patients may not bleed immediately after an injury or minor surgery as they have a normal response with platelet activation and formation of a platelet plug. At times, the diagnosis of hemophilia is not made in these patients until after their first minor procedure (e.g., tooth extraction or tonsillectomy).
Patients with hemophilia A or B are treated with factor VIII or factor IX concentrate, respectively. Recombinant factor VIII is strongly recommended for patients not treated previously and is generally recommended for patients who are both human immunodeficiency virus (HIV) and hepatitis C virus (HCV) seronegative. For factor IX replacement, the preferred products are recombinant or high-purity factor IX. In general, activity levels should be restored to 30% to 40% for mild hemorrhage, 50% for severe bleeding, and 80% to 100% for life-threatening bleeding. Up to 20% of hemophiliacs with factor VIII deficiency develop inhibitors that can neutralize FVIII. For patients with low titers, inhibitors can be overcome with higher doses of factor VIII. For patients with high titer inhibitors, alternate treatments should be used and may include porcine factor VIII, prothrombin complex concentrates, activated prothrombin complex concentrates, or recombinant factor VIIa. For patients undergoing elective surgical procedures, a multidisciplinary approach with preoperative planning and replacement is recommended.2
von Willebrand’s disease (vWD), the most common congenital bleeding disorder, is characterized by a quantitative or qualitative defect in vWF, a large glycoprotein responsible for carrying factor VIII and platelet adhesion. The latter is important for normal platelet adhesion to exposed subendothelium and for aggregation under high shear conditions. Patients with vWD have bleeding that is characteristic of platelet disorders such as easy bruising and mucosal bleeding. Menorrhagia is common in women. vWD is classified into three types. Type I is a partial quantitative deficiency, type II is a qualitative defect, and type III is total deficiency. For bleeding, type I patients usually respond well to desmopressin (DDAVP). Type II patients may respond, depending on the particular defect. Type III patients are usually unresponsive. These patients may require vWF concentrates.3
Factor XI deficiency, an autosomal recessive inherited condition sometimes referred to as hemophilia C, is more prevalent in the Ashkenazi Jewish population but found in all races. Spontaneous bleeding is rare, but bleeding may occur after surgery, trauma, or invasive procedures. Treatment of patients with factor XI deficiency who present with bleeding or in whom surgery is planned and who are known to have bled previously is with fresh frozen plasma (FFP). Each milliliter of plasma contains 1 unit of factor XI activity, so the volume needed depends on the patient’s baseline level, the desired level, and the plasma volume. Antifibrinolytics may be useful in patients with menorrhagia. Factor VIIa is recommended for patients with anti-factor XI antibodies, although thrombosis has been reported.4 There has been renewed interest in factor XI inhibitors as antithrombotic agents, because patients with factor XI deficiency generally have only minimal bleeding risk unless a severe deficiency is present and seem to be protected from thrombosis.5
Inherited deficiencies of factors II, V, and X are rare. These deficiencies are inherited as autosomal recessive. Significant bleeding in homozygotes with less than 1% of normal activity is encountered. Bleeding with any of these deficiencies is treated with FFP. Similar to factor XI, FFP contains one unit of activity of each per milliliter. However, factor V activity is decreased because of its inherent instability. The half-life of prothrombin (factor II) is long (approximately 72 hours), and only about 25% of a normal level is needed for hemostasis. Prothrombin complex concentrates can be used to treat deficiencies of prothrombin or factor X. Daily infusions of FFP are used to treat bleeding in factor V deficiency, with a goal of 20% to 25% activity. Factor V deficiency may be coinherited with factor VIII deficiency. Treatment of bleeding in individuals with the combined deficiency requires factor VIII concentrate and FFP. Some patients with factor V deficiency are also lacking the factor V normally present in platelets and may need platelet transfusions as well as FFP.
Inherited factor VII deficiency is a rare autosomal recessive disorder. Clinical bleeding can vary widely and does not always correlate with the level of FVII coagulant activity in plasma. Bleeding is uncommon unless the level is less than 3%. The most common bleeding manifestations involve easy bruising and mucosal bleeding, particularly epistaxis or oral mucosal bleeding. Postoperative bleeding is also common, reported in 30% of surgical procedures.6 Treatment is with FFP or recombinant factor VIIa. The half-life of recombinant factor VIIa is only approximately 2 hours, but excellent hemostasis can be achieved with frequent infusions. The half-life of factor VII in FFP is up to 4 hours.
Congenital factor XIII (FXIII) deficiency, originally recognized by Duckert in 1960, is a rare autosomal recessive disease usually associated with a severe bleeding diathesis.7 The male-to-female ratio is 1:1. Although acquired FXIII deficiency has been described in association with hepatic failure, inflammatory bowel disease, and myeloid leukemia, the only significant association with bleeding in children is the inherited deficiency.8 Bleeding is typically delayed because clots form normally but are susceptible to fibrinolysis. Umbilical stump bleeding is characteristic, and there is a high risk of intracranial bleeding. Spontaneous abortion is usual in women with factor XIII deficiency unless they receive replacement therapy. Replacement can be accomplished with FFP, cryoprecipitate, or a factor XIII concentrate. Levels of 1% to 2% are usually adequate for hemostasis.
Inherited platelet functional defects include abnormalities of platelet surface proteins, abnormalities of platelet granules, and enzyme defects. The major surface protein abnormalities are thrombasthenia and Bernard-Soulier syndrome. Thrombasthenia, or Glanzmann thrombasthenia, is a rare genetic platelet disorder, inherited in an autosomal recessive pattern, in which the platelet glycoprotein IIb/IIIa (GP IIb/IIIa) complex is either lacking or present but dysfunctional. This defect leads to faulty platelet aggregation and subsequent bleeding. The disorder was first described by Dr. Eduard Glanzmann in 1918.9 Bleeding in thrombasthenic patients must be treated with platelet transfusions. The Bernard-Soulier syndrome is caused by a defect in the GP Ib/IX/V receptor for vWF, which is necessary for platelet adhesion to the subendothelium. Transfusion of normal platelets is required for bleeding in these patients.
The most common intrinsic platelet defect is storage pool disease. It involves loss of dense granules (storage sites for ADP, adenosine triphosphate [ATP], Ca2+, and inorganic phosphate) and α-granules. Dense granule deficiency is the most prevalent of these. It may be an isolated defect or occur with partial albinism in the Hermansky-Pudlak syndrome. Bleeding is variable, depending on the severity of the granule defect. Bleeding is caused by the decreased release of ADP from these platelets. A few patients have been reported who have decreased numbers of both dense and α-granules. They have a more severe bleeding disorder. Patients with mild bleeding as a consequence of a form of storage pool disease can be treated with DDAVP. It is likely that the high levels of vWF in the plasma after DDAVP somehow compensate for the intrinsic platelet defect. With more severe bleeding, platelet transfusion is required.
ACQUIRED HEMOSTATIC DEFECTS
Acquired abnormalities of platelets are much more common than acquired defects and may be quantitative or qualitative, although some patients have both types of defects. Quantitative defects may be a result of failure of production, shortened survival, or sequestration. Failure of production is generally a result of bone marrow disorders such as leukemia, myelodysplastic syndrome, severe vitamin B12 or folate deficiency, chemotherapeutic drugs, radiation, acute ethanol intoxication, or viral infection. If a quantitative abnormality exists and treatment is indicated either due to symptoms or the need for an invasive procedure, platelet transfusion is utilized. The etiologies of both qualitative and quantitative defects are reviewed in Table 4-1.
|
Shortened platelet survival is seen in immune thrombocytopenia, disseminated intravascular coagulation, or disorders characterized by platelet thrombi such as thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Immune thrombocytopenia may be idiopathic or associated with other autoimmune disorders or low-grade B-cell malignancies, and it may also be secondary to viral infections (including HIV) or drugs. Secondary immune thrombocytopenia often presents with a very low platelet count, petechiae and purpura, and epistaxis. Large platelets are seen on peripheral smear. Initial treatment consists of corticosteroids, intravenous gamma globulin, or anti-D immunoglobulin in patients who are Rh positive. Both gamma globulin and anti-D immunoglobulin are rapid in onset. Platelet transfusions are not usually needed unless central nervous system bleeding or active bleeding from other sites occurs. Survival of the transfused platelets is usually short.
Primary immune thrombocytopenia is also known as idiopathic thrombocytopenic purpura (ITP). In children, it is usually acute in onset, short lived, and typically follows a viral illness. In contrast, ITP in adults is gradual in onset, chronic in nature, and has no identifiable cause. Because the circulating platelets in ITP are young and functional, bleeding is less for a given platelet count than when there is failure of platelet production. The pathophysiology of ITP is believed to involve both impaired platelet production and T cell–mediated platelet destruction.10 Management options are summarized in Table 4-2.11 Treatment of drug-induced immune thrombocytopenia may simply entail withdrawal of the offending drug, but corticosteroids, gamma globulin, and anti-D immunoglobulin may hasten recovery of the count. Heparin-induced thrombocytopenia (HIT) is a form of drug-induced immune thrombocytopenia. It is an immunologic event during which antibodies against platelet factor 4 (PF4) formed during exposure to heparin affect platelet activation and endothelial function with resultant thrombocytopenia and intravascular thrombosis.12 The platelet count typically begins to fall 5 to 7 days after heparin has been started, but if it is a re-exposure, the decrease in count may occur within 1 to 2 days. HIT should be suspected if the platelet count falls to less than 100,000 or if it drops by 50% from baseline in a patient receiving heparin. While HIT is more common with full-dose unfractionated heparin (1%–3%), it can also occur with prophylactic doses or with low molecular weight heparins. Interestingly, approximately 17% of patients receiving unfractionated heparin and 8% receiving low molecular weight heparin develop antibodies against PF4, yet a much smaller percentage develop thrombocytopenia and even fewer develop clinical HIT.13 In addition to the mild to moderate thrombocytopenia, this disorder is characterized by a high incidence of thrombosis that may be arterial or venous. Importantly, the absence of thrombocytopenia in these patients does not preclude the diagnosis of HIT.
|
The diagnosis of HIT may be made by using either a serotonin release assay (SRA) or an enzyme-linked immunosorbent assay (ELISA). The SRA is highly specific but not sensitive, so a positive test supports the diagnosis but a negative test does not exclude HIT.12 On the other hand, the ELISA has a low specificity, so although a positive ELISA confirms the presence of anti-heparin-PF4, it does not help in the diagnosis of clinical HIT. A negative ELISA, however, essentially rules out HIT.
The initial treatment of suspected HIT is to stop heparin and begin an alternative anticoagulant. Stopping heparin without addition of another anticoagulant is not adequate to prevent thrombosis in this setting. Alternative anticoagulants are primarily thrombin inhibitors. The most recent guideline by the American College of Chest Physicians recommends lepirudin, argatroban, or danaparoid for patients with normal renal function and argatroban for patients with renal insufficiency.14 Because of warfarin’s early induction of a hypercoagulable state, warfarin should be instituted only once full anticoagulation with an alternative agent has been accomplished and the platelet count has begun to recover.
These are also disorders in which thrombocytopenia is a result of platelet activation and formation of platelet thrombi. In thrombotic thrombocytopenic purpura (TTP), large vWF molecules interact with platelets, leading to activation. These large molecules result from inhibition of a metalloproteinase enzyme, ADAMtS13, which cleaves the large vWF molecules.15 TTP is classically characterized by thrombocytopenia, microangiopathic hemolytic anemia, fever, and renal and neurologic signs or symptoms. The finding of schistocytes on a peripheral blood smear aids in the diagnosis. Plasma exchange with replacement of FFP is the treatment for acute TTP.16 Additionally, rituximab, a monoclonal antibody against the CD20 protein on B lymphocytes, has shown promise as an immunomodulatory therapy directed against patients with acquired TTP, of which the majority are autoimmune mediated.17
Hemolytic uremic syndrome (HUS) often occurs secondary to infection by Escherichia coli 0157:H7 or other Shiga toxin-producing bacteria. The metalloproteinase is normal in these cases. HUS is usually associated with some degree of renal failure, with many patients requiring renal replacement therapy. Neurologic symptoms are less frequent. A number of patients develop features of both TTP and HUS. This may occur with autoimmune diseases, especially systemic lupus erythematosus and HIV infection, or in association with certain drugs (such as ticlopidine, mitomycin C, gemcitabine) or immunosuppressive agents (such as cyclosporine and tacrolimus). Discontinuation of the involved drug is the mainstay of therapy. Plasmapheresis is frequently used, but it is not clear what etiologic factor is being removed by the pheresis.
Sequestration is another important cause of thrombocytopenia and usually involves trapping of platelets in an enlarged spleen typically related to portal hypertension, sarcoid, lymphoma, or Gaucher’s disease. The total body platelet mass is essentially normal in patients with hypersplenism, but a much larger fraction of the platelets are in the enlarged spleen. Platelet survival is mildly decreased. Bleeding is less than anticipated from the count because sequestered platelets can be mobilized to some extent and enter the circulation. Platelet transfusion does not increase the platelet count as much as it would in a normal person because the transfused platelets are similarly sequestered in the spleen. Splenectomy is not indicated to correct the thrombocytopenia of hypersplenism caused by portal hypertension.
Thrombocytopenia is the most common abnormality of hemostasis that results in bleeding in the surgical patient. The patient may have a reduced platelet count as a result of a variety of disease processes, as discussed earlier. In these circumstances, the marrow usually demonstrates a normal or increased number of megakaryocytes. By contrast, when thrombocytopenia occurs in patients with leukemia or uremia and in patients on cytotoxic therapy, there are generally a reduced number of megakaryocytes in the marrow. Thrombocytopenia also occurs in surgical patients as a result of massive blood loss with product replacement deficient in platelets. Thrombocytopenia may also be induced by heparin administration during cardiac and vascular cases, as in the case of HIT, or may be associated with thrombotic and hemorrhagic complications. When thrombocytopenia is present in a patient for whom an elective operation is being considered, management is contingent upon the extent and cause of platelet reduction. A count of greater than 50,000/μL generally requires no specific therapy.
Early platelet administration has now become part of massive transfusion protocols.18,19 Platelets are also administered preoperatively to rapidly increase the platelet count in surgical patients with underlying thrombocytopenia. One unit of platelet concentrate contains approximately 5.5 × 1010 platelets and would be expected to increase the circulating platelet count by about 10,000/μL in the average 70-kg person. Fever, infection, hepatosplenomegaly, and the presence of antiplatelet alloantibodies decrease the effectiveness of platelet transfusions. In patients refractory to standard platelet transfusion, the use of human leukocyte antigen (HLA)-compatible platelets coupled with special processors has proved effective.
Impaired platelet function often accompanies thrombocytopenia but may also occur in the presence of a normal platelet count. The importance of this is obvious when one considers that 80% of overall strength is related to platelet function. The life span of platelets ranges from 7 to 10 days, placing them at increased risk for impairment by medical disorders and prescription and over-the-counter medications. Impairment of ADP-stimulated aggregation occurs with massive transfusion of blood products. Uremia may be associated with increased bleeding time and impaired aggregation. Defective aggregation and platelet dysfunction are also seen in patients with thrombocythemia, polycythemia vera, and myelofibrosis.
Drugs that interfere with platelet function include aspirin, clopidogrel, prasugrel, dipyridamole, and GP IIb/IIIa inhibitors. Aspirin, clopidogrel, and prasugrel all irreversibly inhibit platelet function. Clopidogrel and prasugrel do so through selective irreversible inhibition of ADP-induced platelet aggregation.20 Aspirin works through irreversible acetylation of platelet prostaglandin synthase.
There are no prospective randomized trials in general surgical patients to guide the timing of surgery in patients on aspirin, clopidogrel, or prasugrel.21 The general recommendation is that approximately 5 to 7 days should pass from the time the drug is stopped until an elective procedure is performed.22 Timing of urgent and emergent surgeries is even more unclear. Preoperative platelet transfusions may be beneficial, but there are no good data to guide their administration. However, new functional tests are becoming available that may better demonstrate defects in platelet function and may serve to guide the timing of operation or when platelet transfusions might be indicated.
Other disorders associated with abnormal platelet function include uremia, myeloproliferative disorders, monoclonal gammopathies, and liver disease. In the surgical patient, platelet dysfunction of uremia can often be corrected by dialysis or the administration of DDAVP. Platelet transfusion may not be helpful if the patient is uremic when the platelets are given and only serve to increase antibodies. Platelet dysfunction in myeloproliferative disorders is intrinsic to the platelets and usually improves if the platelet count can be reduced to normal with chemotherapy. If possible, surgery should be delayed until the count has been decreased. These patients are at risk for both bleeding and thrombosis. Platelet dysfunction in patients with monoclonal gammopathies is a result of interaction of the monoclonal protein with platelets. Treatment with chemotherapy or, occasionally, plasmapheresis to lower the amount of monoclonal protein improves hemostasis.
DIC is an acquired syndrome characterized by systemic activation of coagulation pathways that result in excessive thrombin generation and the diffuse formation of microthrombi. This disturbance ultimately leads to consumption and depletion of platelets and coagulation factors with the resultant classic picture of diffuse bleeding. Fibrin thrombi developing in the microcirculation may cause microvascular ischemia and subsequent end-organ failure if severe. There are many different conditions that predispose a patient to DIC, and the presence of an underlying condition is required for the diagnosis. For example, injuries resulting in embolization of materials such as brain matter, bone marrow, or amniotic fluid can act as potent thromboplastins that activate the DIC cascade.23 Additional etiologies include malignancy, organ injury (such as severe pancreatitis), liver failure, certain vascular abnormalities (such as large aneurysms), snake bites, illicit drugs, transfusion reactions, transplant rejection, and sepsis.24 In fact, DIC frequently accompanies sepsis and may be associated with multiple organ failure. As of yet, scoring systems for organ failure do not routinely incorporate DIC. The important interplay between sepsis and coagulation abnormalities was demonstrated by Dhainaut et al who showed that activated protein C was effective in septic patients with DIC.25 The diagnosis of DIC is made based on an inciting etiology with associated thrombocytopenia, prolongation of the prothrombin time, a low fibrinogen level, and elevated fibrin markers (FDPs, D-dimer, soluble fibrin monomers). A scoring system developed by the International Society for Thrombosis and Hemostasis has been shown to have high sensitivity and specificity for diagnosing DIC as well as a strong correlation between an increasing DIC score and mortality, especially in patients with infections.26
The most important facets of treatment are relieving the patient’s causative primary medical or surgical problem and maintaining adequate perfusion. If there is active bleeding, hemostatic factors should be replaced with FFP, which is usually sufficient to correct the hypofibrinogenemia, although cryoprecipitate, fibrinogen concentrates, or platelet concentrates may also be needed. Given the formation of microthrombi in DIC, heparin therapy has also been proposed. Most studies, however, have shown that heparin is not helpful in acute forms of DIC, but may be indicated in cases where thrombosis predominates, such as arterial or venous thromboembolism and severe purpura fulminans.
An acquired hypofibrinogenic state in the surgical patient can be a result of pathologic fibrinolysis. This may occur in patients following prostate resection when urokinase is released during surgical manipulation of the prostate or in patients undergoing extracorporeal bypass. The severity of fibrinolytic bleeding is dependent on the concentration of breakdown products in the circulation. Antifibrinolytic agents, such as ε-aminocaproic acid and tranexamic acid, interfere with fibrinolysis by inhibiting plasminogen activation.
Polycythemia, or an excess of red blood cells, places surgical patients at risk. Spontaneous thrombosis is a complication of polycythemia vera, a myeloproliferative neoplasm, and can be explained in part by increased blood viscosity, increased platelet count, and an increased tendency toward stasis. Paradoxically, a significant tendency toward spontaneous hemorrhage also is noted in these patients. Thrombocytosis can be reduced by the administration of low-dose aspirin, phlebotomy, and hydroxyurea.27
The liver plays a key role in hemostasis because it is responsible for the synthesis of many of the coagulation factors (Table 4-3). Patients with liver disease, therefore, have decreased production of several key non-endothelial cell-derived coagulation factors as well as natural anticoagulant proteins, causing a disturbance in the balance between procoagulant and anticoagulant pathways. This disturbance in coagulation mechanisms causes a complex paradigm of both increased bleeding risk and increased thrombotic risk. The most common coagulation abnormalities associated with liver dysfunction are thrombocytopenia and impaired humoral coagulation function manifested as prolongation of the prothrombin time and international normalized ratio (INR). The etiology of thrombocytopenia in patients with liver disease is typically related to hypersplenism, reduced production of thrombopoietin, and immune-mediated destruction of platelets. The total body platelet mass is often normal in patients with hypersplenism, but a much larger fraction of the platelets is sequestered in the enlarged spleen. Bleeding may be less than anticipated because sequestered platelets can be mobilized to some extent and enter the circulation. Thrombopoietin, the primary stimulus for thrombopoiesis, may be responsible for some cases of thrombocytopenia in cirrhotic patients, although its role is not well delineated. Finally, immune-mediated thrombocytopenia may also occur in cirrhotics, especially those with hepatitis C and primary biliary cirrhosis.28 In addition to thrombocytopenia, these patients also exhibit platelet dysfunction via defective interactions between platelets and the endothelium, and possibly due to uremia and changes in endothelial function in the setting of concomitant renal insufficiency. Hypocoagulopathy is further exacerbated with low platelet counts because platelets help facilitate thrombin generation by assembling coagulation factors on their surfaces. In conditions mimicking intravascular flow, low hematocrit and low platelet counts contributed to decreased adhesion of platelets to endothelial cells, although increased vWF, a common finding in cirrhotic patients, may offset this change in patients with cirrhosis.29 Hypercoagulability of liver disease has recently gained increased attention, with more evidence demonstrating the increased incidence of thromboembolism despite thrombocytopenia and a hypocoagulable state on conventional blood tests.30,31 This is attributed to decreased production of liver-synthesized proteins C and S, antithrombin, and plasminogen levels, as well as elevated levels of endothelial-derived vWF and factor VIII, a potent driver of thrombin generation.32,33 Given the concomitant hypo- and hypercoagulable features seen in patients with liver disease, conventional coagulation tests may be difficult to interpret, and alternative tests such as thromboelastography (TEG) may be more informative of the functional status of clot formation and stability in cirrhotic patients. Several studies imply that TEG provides a better assessment of bleeding risk than standard tests of hemostasis in patients with liver disease; however, no studies have directly tested this, and future prospective trials are needed.34
Before instituting any therapy to ameliorate thrombocytopenia, the actual need for correction should be strongly considered. In general, correction based solely on a low platelet count should be discouraged. Most often, treatment should be withheld for invasive procedures and surgery. Platelet transfusions are the mainstay of therapy; however, the effect typically lasts only several hours. Risks associated with transfusions in general and the development of antiplatelet antibodies in a patient population likely to need recurrent correction should be considered. A potential alternative strategy involves administration of interleukin-11 (IL-11), a cytokine that stimulates proliferation of hematopoietic stem cells and megakaryocyte progenitors.26
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


