Glioblastoma
Alexandros D. Polydorides, MD, PhD
Key Facts
Terminology
Highly malignant (grade IV) astrocytic neoplasm
Etiology/Pathogenesis
Secondary GBM: From lower grade astrocytoma
In 4-5 years; women, children; TP53 mutation
Primary GBM (90%): No evidence of precursor lesion
Short history; older patients; EGFR amplification
Clinical Issues
Most common intracranial neoplasm (12-15%)
Peak: 45-70 years old (70%); ˜ 9% in children
Cerebral hemispheres: Frontal, temporal, parietal
Abrupt onset, rapid/relentless symptom progression
Expansile neoplasm; poor prognosis (3-9 months)
Rapid infiltration of CNS along white matter fibers
Debulking/palliative surgery, chemoradiation
Image Findings
Irregular ring enhancement around central necrosis
Broad, ill-defined peripheral edema/infiltrating tumor
Macroscopic Features
Poorly demarcated; irregular infiltration of cortex
Red-brown hemorrhage, central yellow necrosis
Microscopic Pathology
Poorly differentiated, highly anaplastic astrocytes
Nuclear pleomorphism, brisk mitotic activity
Subpial, subependymal, perineuronal aggregates
Vascular proliferation, pseudopalisading necrosis
1 or both generally required for diagnosis
Perivascular lymphocytes, metaplastic areas seen
Variants: Small cell, giant cell, gliosarcoma
GFAP(+) cells; MIB-1 index > 10%
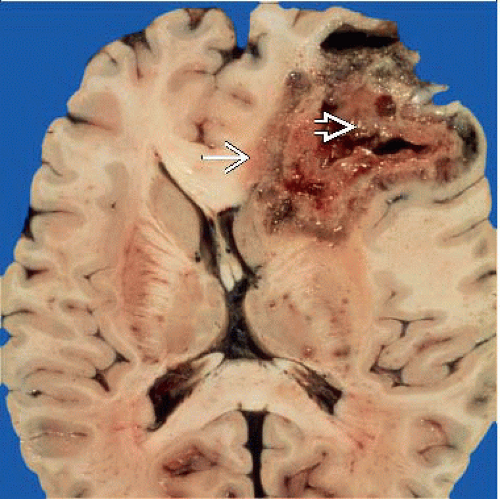 Characteristic heterogeneous gross appearance of GBM: Central necrosis  surrounded by hemorrhage, edema, and infiltrating tumor surrounded by hemorrhage, edema, and infiltrating tumor  . (Courtesy R. Hewlett, MD.) . (Courtesy R. Hewlett, MD.) |
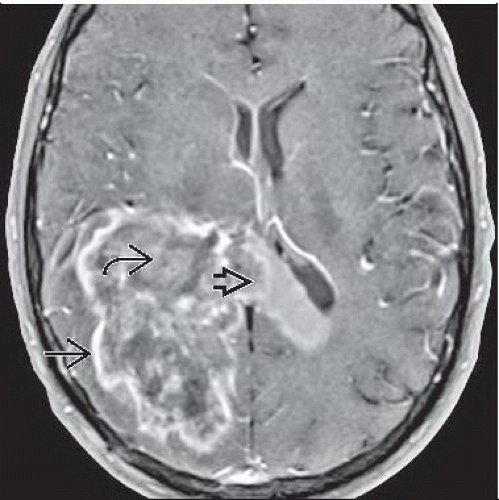 Axial T1 C+ MRI shows peripheral ring enhancement  , central necrosis , central necrosis  , and extension across the splenium of the corpus callosum , and extension across the splenium of the corpus callosum  , all characteristic of GBM. , all characteristic of GBM. |
TERMINOLOGY
Synonyms
Glioblastoma (spongioblastoma) multiforme (GBM)
High-grade (malignant) astrocytoma (WHO grade IV)
Definitions
Highly malignant (WHO grade IV) astrocytic neoplasm mostly of cerebral hemispheres, typically in adults
Fast-growing, poorly differentiated hypercellular tumor with mitoses, microvascular proliferation, and necrosis
ETIOLOGY/PATHOGENESIS
Secondary GBM (Type I or “Progressive”)
Evolves/progresses from lower grade astrocytoma
Diffuse (grade II) or anaplastic (grade III)
Mean interval of 4-5 years
Temporal/spatial association
Lower grade areas coexist, often multiple
Oligodendroglioma may also progress to GBM
Sequential accumulation of genetic alterations
Early and frequent TP53 mutation (> 70%)
Rare EGFR amplification/overexpression (< 10%)
PDGFR overexpression; LOH 10, 17p, 19q
More common in women, younger patients, children
Primary GBM (Type II or “De Novo”)
No evidence of less malignant precursor lesion
Shorter clinical history (< 3 months)
Densely cellular, homogeneously anaplastic
Lower grade lesion possibly overrun/obscured
Distinctly different genetic mutations
EGFR amplification or overexpression (> 50%)
Less common P53 mutation (10%)
PTEN mutation, CDNK2A (p16) deletion
Older patients; represents most GBMs (90%)
Pediatric High-Grade Astrocytomas
Common TP53 mutation, rare EGFR amplification
Especially brainstem lesions
Some evidence of DNA mismatch repair
Resulting microsatellite instability (MSI)
Genetic Predisposition/Familial Syndromes
Turcot syndrome, Maffucci syndrome, NF1
Multiple enchondromatosis type I (Ollier disease)
Germline TP53 mutations (Li-Fraumeni syndrome)
Prior Ionizing Radiation
Only known environmental risk for high-grade glioma
Considered a contributing factor in < 1% of patients
Site of GBM needs to be within prior irradiation field
Average latency period: 10-15 years
Rare Unproven Associations
Meningioma, AIDS, demyelinating diseases
Multifocal Glioblastoma
Independent, polyclonal lesions proven in 3-5% cases
Usually limited to inherited predisposing syndromes
True multicentricity/multifocality difficult to measure
May be from widespread extension (satellite lesions)
CLINICAL ISSUES
Epidemiology
Incidence
2-3 cases per 100, 000 people per year
Most common intracranial neoplasm (12-15%)
5-10% of childhood intracranial neoplasms
Most common astrocytic tumor (50-70%)
Age
Any age; peak: 45-70 years (70%)
˜ 9% in children, rare congenital cases
Gender
Slight male predominance (3:2)
Site
Mostly cerebral hemispheres (subcortical white matter)
Frontal, temporal, parietal > occipital lobes
Brainstem (“malignant brainstem glioma”), thalamus
Mostly in children
May be bilaterally symmetrical or multifocal (20%)
Rare: Cerebellum, optic nerve, spinal cord, ventricles
Presentation
Abrupt onset, rapid/relentless symptom progression
Increased intracranial pressure (life-threatening)
Epileptic seizures, acute intracranial hemorrhage
Nonspecific neurologic symptoms, but may be focal
Mental status/personality changes, headache, nausea
Pons lesions: Long tract/cranial nerve signs, ataxia
Natural History
Aggressive, expansile neoplasm with poor prognosis
Due to lack of localization, late detection
Rapid infiltration/invasion/spread within CNS
Adjacent cortex, basal ganglia, subependymal
Along compact white matter (myelinated) fibers
Corpus callosum to contralateral hemisphere
“Butterfly glioma” (bilateral, symmetric lesion)
Fornix, internal capsule, optic radiation, anterior commissure, perivascular (Virchow-Robin) spaces
May lead to multifocality at presentation
Provide pathway for post-treatment recurrence
Subarachnoid space/CSF seeding in ˜ 10%
Especially after long postoperative period
More common in brain stem, spinal cord lesions
Cranial (dural/skull) extension: Less common
Rare distant systemic spread (hematogenous)
Bone, lymph nodes, liver, lung
Usually after surgical intervention
Multifactorial cause of death
Local effects, progressive deficits, radionecrosis
Pulmonary embolism a common complication
Treatment
Surgical approaches
More successful with better demarcated lesions
Often for debulking or supportive/palliative reasons
Adjuvant therapy
Radiation, chemotherapy are standard treatment
Fail to improve outcome or prevent recurrence
Prognosis
Mean postoperative survival time: 3-9 months
5-year survival rates < 20% in most studies
Reduced survival correlated with
EGFR amplification/overexpression
Loss of chromosome 10; lack of TP53 mutation
MIB-1 index (Ki-67 immunostain) > 7.5%
Brainstem, spinal cord location
Extent of necrosis
Favorable prognostic factors
Young patient age (< 45 years old)
Secondary GBM (vs. primary/de novo)
Larger extent (gross total) resection
High performance status (Karnofsky scale)
Long duration of preoperative symptoms
Presence of better differentiated component
Giant cell GBM, oligodendroglial component
IMAGE FINDINGS
General Features
Best diagnostic clue
Irregular ring enhancement around central necrosis
Location
Usually solitary but may form satellite lesions
Primary lesion: Deep seated; satellites: Superficial
MR Findings
T1WI: Isointense to hypointense, necrotic, cystic mass
Irregular, thick rim of contrast enhancement
Corresponds to highly cellular, vascular neoplasm
Infiltrating glioma extends beyond enhancing rim
T2WI/FLAIR: Heterogeneous, hyperintense mass
Broad, ill-defined peripheral area of low attenuation
Corresponds to edema and infiltrating tumor
CT Findings
Irregularly shaped, iso-/hypodense, expansile mass
Peripheral ring-like contrast enhancement
Dark, hypodense central area of necrosis
Marked mass effect, surrounding edema
PET
Malignant tumors: ↑ cellularity, ↑ glucose metabolism
GBM: ↑ FDG uptake; also correlates with ↓ survival
Imaging Findings for Giant Cell GBM
Discrete, subcortical solid mass; lacks central necrosis
Homogeneous enhancement: Resembles metastasis
MACROSCOPIC FEATURES
General Features
Generally ill defined, poorly demarcated
Irregular infiltration of cortex, expansion of gyri
Appears better demarcated than WHO grade II/III
Discrete extension into subarachnoid, meninges
Desmoplastic, collagen-rich reaction
May resemble extraaxial meningioma or metastasis
Red-brown areas of recent and remote hemorrhage
Usually small, multiple, and diffuse
May form large thrombosed hypervascular mass
Central yellow necrotic areas with myelin breakdown
May involve up to 80-90% of tumor mass
Liquefactive necrosis can lead to macroscopic cysts
Sections to Be Submitted
Generously sample tumor, including
Nonnecrotic, gray, fleshy, viable areas
To achieve diagnostic histology
Less dense-appearing peripheral areas
To identify precursor glioma, if present
Necrotic, hemorrhagic areas
To find pseudopalisading, vascular proliferation
Size
Often very large, occupying much of lobe
Giant Cell GBM and Gliosarcoma
May be well demarcated, firm, resemble metastasis
MICROSCOPIC PATHOLOGY
Histologic Features
Variable histologic appearance (“multiforme”)
Loose aggregates or cellular sheets of neoplastic cells
Prominent intersecting bundles or fascicles
More heterogeneity seen in secondary GBM
In terms of size, fibrillarity, process formation
Lower grade areas (WHO II, III) may coexist
Abrupt or continuous transition to GBM
Architectural organization of tumor in zones
Central regions of coagulative necrosis
Corresponds to radiologic dark hypodense area
Surrounded by rims of densely cellular tissue
Correspond to radiologic contrast enhancement
Peripheral tumor cells infiltrating brain parenchyma
Correspond to surrounding radiologic edema
Prominent “secondary structures” (of Scherer)
Perivascular/subpial/subependymal aggregates
Perineuronal satellitosis
Fusiform tumor cells within myelinated pathways
Especially in areas of infiltration into cortex
Interaction of neoplastic cells with native brain
Microvascular (endothelial) proliferation
Glomeruloid capillary tufts (microvascular)
Multilayered, proliferating endothelial cells
May also include smooth muscle cells, pericytes
Usually along edge of necrotic, ischemic tumor
Glioma-secreted angiogenic substances (VEGF)
Vascular thrombosis often present
2nd form of vascular hyperplasia
Medium-sized vessels (not truly “microvascular”)
Intraluminal endothelial cell proliferation
Less common, poorer prognosis
Geographic tumor necrosis
Small, irregular, band-like, or serpiginous areas
Outlined by “pseudopalisading” pattern
Radially oriented, small, fusiform tumor cells
Central area of necrotic fibrillary network
Probably secondary to hypoxia-induced apoptosis
Coalesce into larger necrotic areas
Necrotic tumor cells, vessels
Eventually lack pseudopalisading
Often in primary GBM, indicate poorer prognosis
Correspond to radiologic nonenhancing areas
Ischemic, due to insufficient blood supply
Macrophages not prominent
Perivascular lymphocyte collections
Mostly CD8(+) T cells, fewer CD4(+) T cells
Some B cells, rare plasma cells
Especially in gemistocytic or giant cell areas
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


