Familial Cancer Syndromes
Satish K. Tickoo, MD
Victor E. Reuter, MD
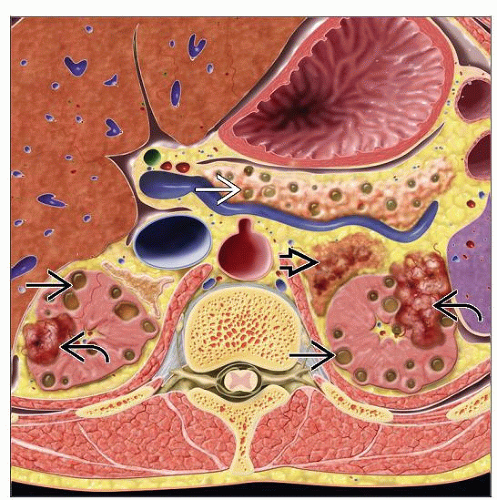 Graphic representation of abdominal lesions in von Hippel-Lindau syndrome shows bilateral, multiple renal cysts  , renal tumors , renal tumors  , pancreatic cysts , pancreatic cysts  , and adrenal pheochromocytoma , and adrenal pheochromocytoma  . . |
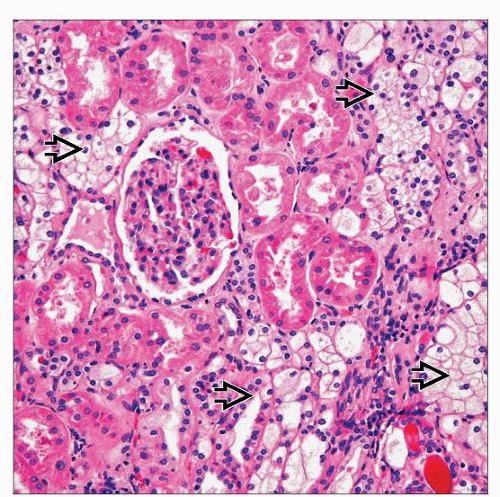 Besides the larger, macroscopic clear cell renal cell carcinomas, VHL kidneys often show numerous microscopic aggregates or tumorlets of clear cells, frequently with irregular outlines  . . |
TERMINOLOGY
Abbreviations
von Hippel-Lindau (VHL) syndrome
Hereditary papillary renal carcinoma (HPRC) syndrome
Birt-Hogg-Dubé (BHD) syndrome
Hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome
Tuberous sclerosis (TS) syndrome
SYNDROMES
General Considerations
In all forms of inherited renal neoplasms, tumors are usually diagnosed at earlier age and are more likely to be multifocal and bilateral
At present, only exception to multifocality and bilaterality appears to be HLRCC syndrome
Because of consistent defects within tumor groups, genetic profiles of inherited neoplasms are relatively easier to study
Knowledge thus gained may be applied in similar, more common sporadic tumors
This has resulted in better understanding of genetic mechanisms involved in various sporadic tumor subtypes
Most targeted therapies currently in use/under investigation have been direct consequence of this better understanding of tumor genetics
SYNDROMIC RENAL TUMORS
von Hippel-Lindau Syndrome
Autosomal dominant syndrome, characterized by
Retinal hemangioblastomas
Clear cell renal cell carcinomas (RCC) and multiple renal cysts
Cerebellar and spinal hemangioblastomas
Pheochromocytomas
Pancreatic cysts and endocrine pancreatic tumors
Endolymphatic sac tumors of ear
Epididymal cystadenomas
VHL, unlike most other familial renal cancer syndromes, shows high degree of genetic penetrance
Estimated incidence: 1/36,000-1/45,500
Syndrome is associated with alterations in tumor suppressor VHL gene
Gene located at chromosome 3p25
Inactivated by various mutations, loss of heterozygosity (LOH), hypermethylations, or alterations in VHL modifier genes
In VHL syndrome, germline mutation present in 1 allele of VHL gene
Clinical manifestations of disease result when mutations/silencing occur in other wild-type allele
VHL gene product pVHL essential for proteosomic degradation of hypoxia-inducible factor-1α (HIF-1α)
Absence of functional pVHL results in overexpression of HIF-1α
Activated HIF-1 heterodimers localize to nucleus and regulate transcription of multiple genes by binding to hypoxia-responsive elements (HRE); activated targets include
Vascular endothelial and platelet-derived growth factors (VEGF and PDGF) and receptors
Glucose transporter protein-1 (GLUT1)
Erythropoietin
Carbonic anhydrase-IX (CA9)
Transforming growth factor-alpha (TGF-α)
C-X-C chemokine receptor type 4 (CXCR4)
C-mesenchymal-epithelial transition factor (c-MET)
Many of these factors associated with angiogenesis, tumorigenesis, and tumor metastasis
Depending on whether pheochromocytomas are present or not, VHL disease can be divided into 2 major types
Type 1 is not associated with pheochromocytomas
It involves “loss of function” mutations, including deletion, microinsertion, and nonsense mutations
Type 2 has high risk for pheochromocytomas and is divided into 3 subtypes
Type 2A, associated with low risk for RCC
Type 2B, associated with high risk for RCC
Type 2C, with pheochromocytomas only
Mutations that predispose to type 2 VHL are mainly of missense type that result in conformationally altered pVHL
These mutant pVHLs still may be able to retain some of their functions or may gain other novel functions
Renal lesions in VHL syndrome include multiple bilateral benign cysts, atypical cysts, cystic RCCs, and solid RCCs
Kidneys are usually of normal size and weight, chiefly because most cysts and RCCs are small
Renal cysts
Cysts are usually few (3-30 in number; mean: 7.8 per kidney), usually small (almost all < 1.5 cm, mean size: 0.7 cm)
Cysts may be unilocular or multilocular
They are almost entirely lined by clear cells; focal or predominant granular cytoplasm is rarely present
Cysts are designated as benign cysts (1 layer of clear cells without atypia) or atypical cysts (2 or 3 cells thick ± atypia)
Focal proliferations more than 3 cells thick are regarded as cystic RCCs
Increased vascularity is often seen around cysts
Clear cell renal cell carcinoma
Mean age for development of renal carcinoma: 37 years (range: 16-67)
By age 70, chance of kidney cancer is 70%
Retinal and CNS hemangioblastomas usually manifest at earlier mean ages (25 and 30 years)
Renal lesions are earlier manifestation in only 7%
In spite of relatively few patients developing metastasis, metastatic RCC is leading cause of death from VHL
In addition to macroscopically identifiable tumors, numerous microscopic nodules of clear cells seen in
VHL kidneys
Some nodules well circumscribed
Others present as aggregates of clear cells, with irregular outlines
Clusters and sheets of clear cells appearing to percolate between nephrons also common
Screening for renal tumors in VHL patients recommended after age 10
Management of renal tumors
Current strategies advocate conservative management for all genetic, multifocal, bilateral tumors
Nephron-sparing surgery/tumor ablation strategy is used with intent to remove all solid and semicystic lesions from kidney
Procedure is usually delayed until tumors grow beyond 3 cm in size
During follow-up, as new tumors develop, repeat procedures are performed
Main intent of this approach is to preserve renal function as much and as long as possible
Targeted therapies currently being investigated to potentially reduce tumor burden of even localized tumors in VHL
Hereditary Papillary Renal Carcinoma Syndrome
Autosomal dominant syndrome, with incomplete penetrance, characterized by
Multiple, bilateral papillary renal cell carcinomas
Hundreds to thousands of tumors known to occur in each kidney
Syndrome is associated with activating mutations of c-MET proto-oncogene
Gene is located at chromosome 7q31
Hepatocyte growth factor (a.k.a. scatter factor) acts as ligand for MET trans-member tyrosine kinase protein
Normally, binding to hepatocyte growth factor activates MET tyrosine kinase protein
Tyrosine phosphorylation induces proliferation and differentiation of epithelial and endothelial cells, cell branching, and invasion
c-MET mutations result in ligand-independent constitutive activation of MET tyrosine kinase
Activated tyrosine kinase then binds to and activates several signal transducers and adaptors, such as
Phosphatidylinositol 3 kinase (PI3K)
pp60src
Growth factor receptor-bound protein 2 (Grb2)
GRB2-associated binding protein 1 (Gab1)
This constitutive activation results in tumorigenesis
Renal tumors associated with syndromic c-MET mutations are all type 1 papillary RCC
Tumors show papillary or tubulo-papillary architecture, similar to type 1 sporadic carcinomas
Foamy macrophages and calcifications commonly present
Tumors often manifest at relatively late age (50 to 70 years)
Recently, early onset form of disease has also been described
Low genetic penetrance is supported by relatively low proportion of cases demonstrating syndrome manifestations
Approximately 50% of members of affected families develop disease
Tumors are multifocal and bilateral
No extrarenal manifestations of HPRC are known at present
Birt-Hogg-Dubé Syndrome
Autosomal dominant syndrome with incomplete penetrance, characterized by
Renal tumors
Cutaneous lesions (fibrofolliculomas, trichodiscomas, and acrochordons)
Pulmonary cysts, spontaneous pneumothorax, bronchiectasis, and bronchospasm
Colonic neoplasms
Medullary thyroid carcinoma
Lipomas
Syndrome involves mutations in BHD gene
BHD gene maps to chromosome 17p12-q11.2
Gene codes for folliculin protein
Multiple mutations, including germline and somatic, have been reported in BHD gene
Usually, germline mutation in 1 allele is inherited, followed by somatic-type mutation in the other allele that may result in tumorigenesis
This supports the role of BHD as a tumor suppressor gene
Renal tumors in BHD syndrome usually have oncocytic cytoplasm
Most common tumor type displays hybrid features of renal oncocytoma and chromophobe RCC
Characteristically, many oncocytic tumors show scattered clusters of cells with clear cytoplasm
Pure chromophobe RCC and renal oncocytomas are other common tumor types
Other types of renal cell carcinoma are also seen, including clear cell and papillary RCC
Renal oncocytosis is evident in surrounding renal parenchyma in large proportion of cases
Morphologic spectrum of renal oncocytosis includes
Numerous microscopic oncocytic nodules: May have features of chromophobe RCC, oncocytoma, or even hybrid tumors
Cysts lined by oncocytic cells
Oncocytic changes in nonneoplastic renal tubules
Clusters and sheets of oncocytic cells percolating between nonneoplastic nephrons
Skin tumors usually appear before renal manifestations
Renal tumors are usually diagnosed in 6th decade of life (range: 31-73 years)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


