Objectives
- Identify the principal hormones secreted from the endocrine pancreas, their cells of origin, and their chemical nature.
- Understand the nutrient, neural, and hormonal mechanisms that regulate pancreatic hormone release.
- List the principal target organs for insulin and glucagon action and their major physiologic effects.
- Identify the time course for the onset and duration of the biologic actions of insulin and glucagon.
- Identify the disease states caused by oversecretion, undersecretion, or decreased sensitivity to insulin, and describe the principal manifestations of each.
Endocrine Pancreas: Introduction
The pancreas is a mixed exocrine and endocrine gland that plays a central role in digestion and in the metabolism, utilization, and storage of energy substrates. This chapter focuses on the endocrine function of the pancreas through the release of insulin and glucagon and the mechanisms by which these hormones regulate events essential to maintaining glucose homeostasis. Maintenance of glucose homeostasis is similar to the maintenance of calcium balance discussed in Chapter 5, in which several tissues and hormones interact in the regulatory process. In the case of glucose, the process involves a regulated balance among hepatic glucose release (from glycogen breakdown and gluconeogenesis), dietary glucose absorption, and glucose uptake and disposal by skeletal muscle and adipose tissue. The pancreatic hormones insulin and glucagon play central roles in regulating each of these processes, and their overall effects are in part modified by other hormones such as growth hormone, cortisol, and epinephrine. In addition to secreting insulin and glucagon, the endocrine pancreas also secretes somatostatin, amylin, and pancreatic polypeptide.
Functional Anatomy
The pancreas is a retroperitoneal gland divided into a head, body, and tail that is located near the duodenum. Most of the pancreatic mass is composed of exocrine cells that are clustered in lobules (acini) divided by connective tissue and connected to a duct that drains into the pancreatic duct and into the duodenum. The product of the pancreatic exocrine cells is an alkaline fluid rich with digestive enzymes, which is secreted into the small intestine to aid in the digestive process. Embedded within the acini are richly vascularized, small clusters of endocrine cells called the islets of Langerhans, in which 2 endocrine cell types (β and α) predominate. The β-cells constitute most of the total mass of endocrine cells, and their principal secretory product is insulin. The α-cells account for approximately 20% of the endocrine cells and are responsible for glucagon secretion. A small number of δ-cells secrete somatostatin, and an even smaller number of cells secrete pancreatic polypeptide. The localization of these cell types within the islets has a particular pattern, with the β-cells located centrally, surrounded by α- and δ-cells.
The arterial blood supply to the pancreas is derived from the splenic artery and the superior and inferior pancreaticoduodenal arteries. Although islets represent only 1%–2% of the mass of the pancreas, they receive approximately 10%–15% of the pancreatic blood flow. The rich vascularization by fenestrated capillaries allows ready access to the circulation for the hormones secreted by the islet cells. Venous blood from the pancreas drains into the hepatic portal vein. Therefore, the liver, a principal target organ for the physiologic effects of pancreatic hormones, is exposed to the highest concentrations of pancreatic hormones. Following first-pass hepatic metabolism, the pancreatic endocrine hormones are distributed to the systemic circulation.
Parasympathetic, sympathetic, and sensory nerves richly innervate the pancreatic islets, and the respective neurotransmitters and neuropeptides released from their nerve terminals exert important regulatory effects on pancreatic endocrine hormone release. Acetylcholine, vasoactive intestinal polypeptide, pituitary adenylate cyclase-activating polypeptide, and gastrin-releasing peptide are released from the parasympathetic nerve terminals. Norepinephrine, galanin, and neuropeptide Y are released from sympathetic nerve terminals. Vagal nerve activation stimulates the secretion of insulin, glucagon, somatostatin, and pancreatic polypeptide. Sympathetic nerve stimulation inhibits basal and glucose-stimulated insulin secretion and somatostatin release and stimulates glucagon and pancreatic polypeptide secretion.
Pancreatic Hormones
The process involved in the synthesis and release of insulin, a polypeptide hormone, by the β-cells of the pancreas is similar to that of other peptide hormones, as discussed in Chapter 1 (Figure 1–2). Preproinsulin undergoes cleavage of its signal peptide during insertion into the endoplasmic reticulum, generating proinsulin (Figure 7–1). Proinsulin consists of an amino-terminal β-chain, a carboxy-terminal α-chain, and a connecting peptide; known as the C-peptide, that links the α- and β-chains. Linking of the 2 chains allows proper folding of the molecule and the formation of disulfide bonds between the 2 chains. In the endoplasmic reticulum, proinsulin is processed by specific endopeptidases, which cleave the C-peptide exposing the end of the insulin chain that interacts with the insulin receptor, generating the mature form of insulin. Insulin and the free C-peptide are packaged into secretory granules in the Golgi. These secretory granules accumulate in the cytoplasm in 2 pools; a readily releasable (5%) and a reserve pool of the granules (more than 95%). On stimulation, the β-cell releases insulin in a biphasic pattern; initially from the readily releasable pool followed by the reserve pool of granules. Only a small proportion of the cellular stores of insulin are released even under maximal stimulatory conditions. Insulin circulates in its free form, has a half-life of 3–8 minutes, and is degraded predominantly by the liver, with more than 50% of insulin degraded during its first pass. Additional degradation of insulin occurs in the kidneys as well as at target tissues by insulin proteases following endocytosis of the receptor-bound hormone.
Figure 7–1.
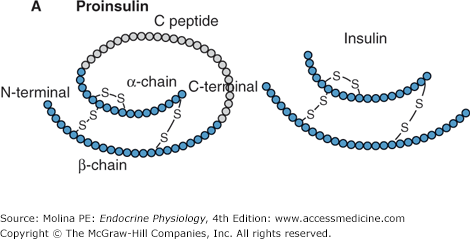
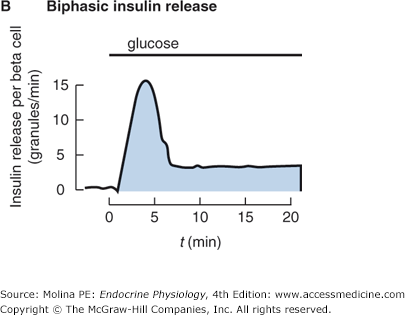
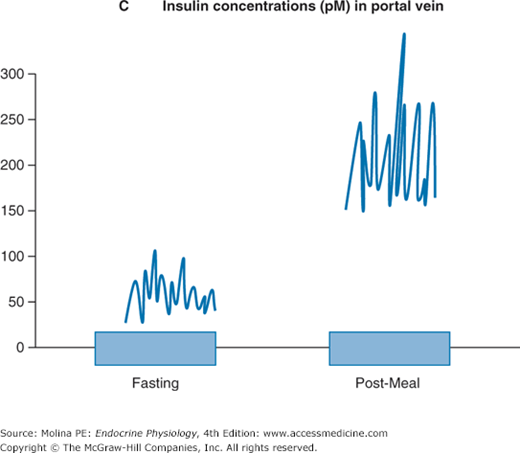
A. Insulin is a peptide hormone synthesized from preproinsulin. Preproinsulin undergoes posttranslational modification in the endoplasmic reticulum (ER) to form proinsulin. The active form of insulin is produced by modification of proinsulin by cleavage of the C-peptide structure linking the alpha and beta chains. Both insulin and the cleaved C-peptide are packaged in secretory granules and are coreleased in response to glucose stimulation. B. Insulin release occurs in a biphasic mode; from readily releasable secretory granules and from granules that must undergo a series of preparatory reactions including mobilization to the plasma membrane. C. In response to a meal, the increase in insulin release results from a higher frequency and amplitude of pulsatile release. Shown are portal insulin concentrations during basal state (left) and after a meal.
Exocytosis of secretory granule content results in the release of equal amounts of insulin and C-peptide into the portal circulation. The importance of C-peptide is that unlike insulin, it is not readily degraded in the liver. Thus, the relatively long half-life of the peptide (35 minutes) allows its release to be used as an index of the secretory capacity of the endocrine pancreas. C-peptide, may have some biologic action as recent evidence indicates that replacement of C-peptide improves renal function and nerve dysfunction in patients with type 1 diabetes. The receptor and signaling mechanisms involved in mediating these responses are still under investigation.
The amino acid sequence of insulin is highly conserved among species. In the past, porcine and bovine insulin were used to treat patients with diabetes. Currently, human recombinant insulin is available and has replaced animal-derived insulin, avoiding problems such as the development of antibodies to nonhuman insulin.
 The pancreatic β-cell functions as a neuroendocrine integrator that responds to changes in plasma levels of energy substrates (glucose and amino acids), hormones (insulin, glucagon-like peptide I, somatostatin, and epinephrine), and neurotransmitters (norepinephrine and acetylcholine) by increasing or decreasing insulin release (Figure 7–2). Glucose is the principal stimulus for insulin release from the pancreatic β-cells. In addition, glucose exerts a permissive effect for the other modulators of insulin secretion.
The pancreatic β-cell functions as a neuroendocrine integrator that responds to changes in plasma levels of energy substrates (glucose and amino acids), hormones (insulin, glucagon-like peptide I, somatostatin, and epinephrine), and neurotransmitters (norepinephrine and acetylcholine) by increasing or decreasing insulin release (Figure 7–2). Glucose is the principal stimulus for insulin release from the pancreatic β-cells. In addition, glucose exerts a permissive effect for the other modulators of insulin secretion.
Figure 7–2.
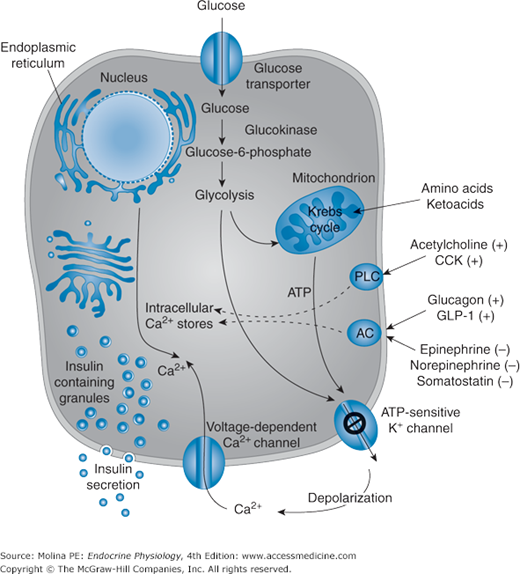
Regulation of insulin release. Glucose is the principal stimulus for insulin release from the pancreatic β-cell. Glucose enters the β-cell cell by a specific glucose transporter protein (GLUT 2) undergoes glycolysis leading to generation of ATP. The increased ATP/ADP ratio leads to inhibition and closure of the ATP-sensitive K+ channels (the target of sulfonylurea drugs), resulting in plasma membrane depolarization and opening of the voltage-dependent Ca2+ channels. The increased Ca2+ influx coupled with mobilization of Ca2+ from intracellular stores leading to the fusion of insulin-containing secretory granules with the plasma membrane and the release of insulin (and C-peptide) into the circulation. Addition factors can also stimulate insulin release from the β-cell, including hormones (glucagon-like peptide 1) and neurotransmitters (acetylcholine). Glucose synergizes with these mediators and enhances the secretory response of the β-cell to these factors. AC, adenylate cyclase; ADP, adenosine diphosphate; ATP, adenosine triphosphate; CCK, cholecystokinin; GLP 1, glucagon-like peptide-1; PLC, phospholipase C. (Modified, with permission, from Fajans SS, Bell GI, Polonsky KS. Mechanisms of disease: molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345:971. Copyright © Massachusetts Medical Society. All rights reserved.)
 The glucose-induced stimulation of insulin release is the result of glucose metabolism by the β-cell (see Figure 7–2). Glucose enters the β-cell through a membrane-bound glucose transporter 2 (GLUT 2) and undergoes immediate phosphorylation by glucokinase in the initial step of glycolysis, leading eventually to the generation of adenosine triphosphate (ATP) by the Krebs cycle. The resulting increase in intracellular ATP to adenosine diphosphate ratio inhibits (closes) the ATP-sensitive K+ channels (KATP) in the β-cell, reducing the efflux of K+. Decreased K+ efflux results in membrane depolarization; activation (opening) of voltage-dependent Ca2+ channels, and increased Ca2+ influx. The increase in intracellular Ca2+ concentrations triggers the exocytosis of insulin secretory granules and the release of insulin into the extracellular space and into the circulation. It is important to note that the regulation of K+ channels by ATP is mediated by the sulfonylurea receptor. This is the basis for the therapeutic use of sulfonylurea drugs in the treatment of diabetes.
The glucose-induced stimulation of insulin release is the result of glucose metabolism by the β-cell (see Figure 7–2). Glucose enters the β-cell through a membrane-bound glucose transporter 2 (GLUT 2) and undergoes immediate phosphorylation by glucokinase in the initial step of glycolysis, leading eventually to the generation of adenosine triphosphate (ATP) by the Krebs cycle. The resulting increase in intracellular ATP to adenosine diphosphate ratio inhibits (closes) the ATP-sensitive K+ channels (KATP) in the β-cell, reducing the efflux of K+. Decreased K+ efflux results in membrane depolarization; activation (opening) of voltage-dependent Ca2+ channels, and increased Ca2+ influx. The increase in intracellular Ca2+ concentrations triggers the exocytosis of insulin secretory granules and the release of insulin into the extracellular space and into the circulation. It is important to note that the regulation of K+ channels by ATP is mediated by the sulfonylurea receptor. This is the basis for the therapeutic use of sulfonylurea drugs in the treatment of diabetes.
The β-cell Ca2+ concentrations can also be elevated by amino acids through their metabolism and ATP generation, or by direct depolarization of the plasma membrane. Other factors (shown in Figure 7–2) that amplify the glucose-induced release of insulin from the β-cell include acetylcholine; cholecystokinin; gastrointestinal peptide, also known as glucose-dependent insulinotropic polypeptide; and glucagon-like peptide 1 (GLP 1). These substances all bind to cell surface receptors and trigger downstream signaling mechanisms controlling insulin release. Acetylcholine and cholecystokinin promote phosphoinositide breakdown, with a consequent mobilization of Ca2+ from intracellular stores, Ca2+ influx across the cell membrane, and activation of protein kinase C. GLP 1 increases levels of cyclic 3′,5′-adenosine monophosphate (cAMP) and activates cAMP-dependent protein kinase A. The generation of cAMP, inositol 1,4,5-trisphosphate, diacylglycerol, and arachidonic acid and the activation of protein kinase C amplify the Ca2+ signal by decreasing Ca2+ uptake by cellular stores and promoting both the phosphorylation and activation of proteins that trigger the exocytosis of insulin. Catecholamines and somatostatin inhibit insulin secretion through G protein–coupled receptor mechanisms, inhibition of adenylate cyclase, and modification of Ca2+ and K+ channel gating.
The short-term regulation of insulin release is mediated through modification of proinsulin mRNA translation. Over longer periods, glucose also increases proinsulin mRNA content by both stimulating proinsulin gene transcription and stabilizing the mRNA. As mentioned above, the release of insulin in response to glucose is biphasic, with an initial rapid release of preformed insulin followed by a more sustained release of newly synthesized insulin. This biphasic response to glucose is a major characteristic of glucose-stimulated insulin secretion. The first phase occurs over a period of minutes, the second over an hour or more. Several hypotheses have been proposed to explain the biphasic nature of insulin secretion; including the involvement of 2 separate pools of insulin granules.
The release of insulin throughout the day is pulsatile and rhythmic in nature (see Figure 7–1). The pulsatile release of insulin is important for achieving maximal physiologic effects. In particular, it appears to be critical in the suppression of liver glucose production and in insulin-mediated glucose disposal by adipose tissue. Insulin release increases after a meal in response to the increases in plasma levels of glucose and amino acids. Secretion is the result of a combination of an increase in the total amount of insulin released in each secretory burst and an increased pulse frequency of a similar magnitude (see Figure 7–1). The synchronized increase in insulin release is thought to be the result of recruitment of β-cells to release insulin. Although it is not clear how the β-cells communicate with each other to synchronize the release of insulin, some of the proposed mechanisms include gap junctions allowing the passage of ions and small molecules; and propagation of membrane depolarization, aiding the synchronization between the cells. In addition, intra-pancreatic neural, hormonal, and substrate factors have been shown to play an important role in the pulsatile pattern of insulin release.
Insulin produces a wide variety of effects that range from immediate (within seconds), such as the modulation of ion (K+) and glucose transport into the cell; early (within minutes), such as the regulation of metabolic enzyme activity; moderate (within minutes to hours), such as the modulation of enzyme synthesis; to delayed (within hours to days), such as the effects on growth and cellular differentiation. Overall, the actions of insulin at target organs are anabolic and promote the synthesis of carbohydrate, fat, and protein, and these effects are mediated through binding to the insulin receptor (Table 7–1).
| Metabolic effects | Insulin stimulates | Insulin inhibits |
|---|---|---|
| Carbohydrate metabolism |
|
|
| Lipid metabolism |
|
|
| Protein metabolism |
|
|
The insulin receptor is part of the insulin-receptor family, which includes the insulin-like growth-factor receptor (Figure 7–3). The insulin receptor is a heterotetrameric glycoprotein membrane receptor composed of 2 α- and 2 β-subunits, linked by disulfide bonds. The extracellular α-chain is the site for insulin binding. The intracellular segment of the β-chain has intrinsic tyrosine kinase activity, which on insulin binding, undergoes autophosphorylation on tyrosine residues. The activated receptor phosphorylates tyrosine residues of several proteins known as insulin receptor substrates 1 through 4 (IRS-1–4); facilitating the interaction of the insulin receptor with intracellular substrates. The result is the coupling of insulin receptor activation to signaling pathways, mainly the phosphatidylinositol 3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK) pathways (see Figure 7–3).
Figure 7–3.
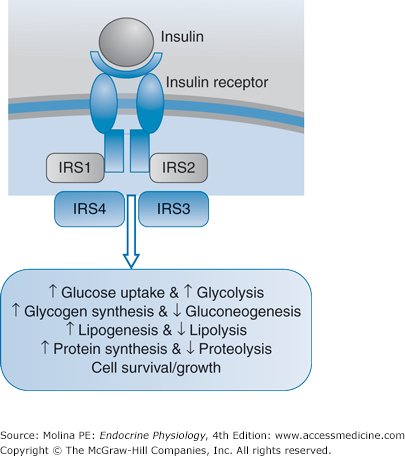
Insulin receptor signaling. Insulin binding to the receptor activates the intrinsic kinase activity of the intracellular domain of the receptor. This results in downstream activation of cellular events mediated through the phosphorylation of insulin receptor substrates (IRS). Downstream signaling pathways involved in insulin-mediated effects including the phosphatidylinositol 3-kinase (PI3K) and the mitogen-activated protein kinase (MAPK) cascades. Activation of phosphoinositide-3 kinase is a major pathway in the mediation of insulin-stimulated glucose transport and metabolism. Among the immediate effects of insulin is the active recruitment of glucose transporter 4 (GLUT 4), stored in intracellular vesicles to the cell surface. Exercise can also stimulate glucose transport by pathways that are independent of phosphoinositide-3 kinase and thought to involve 5′-adenosine monophosphate (AMP)-activated kinase.




