KEY POINTS
Congenital heart disease comprises a wide morphologic spectrum. In general, lesions can be conceptualized as those that can be completely repaired, those that should be palliated, and those that can be either repaired or palliated depending on particular patient and institutional characteristics.
Percutaneous therapies for congenital heart disease are quickly becoming important adjuncts, and in some cases, alternatives, to standard surgical therapy. Important examples include percutaneous closure of atrial and ventricular septal defects, the hybrid approach to hypoplastic left heart syndrome, radiofrequency perforation of the pulmonary valve, and percutaneous pulmonary valve placement. Further studies are necessary to establish criteria and current benchmarks for the safe integration of these novel approaches into the care of patients with congenital heart surgery.
Patients with critical left ventricular outflow tract obstruction, such as neonatal critical aortic stenosis, represent a challenging population. It is critical that the correct decision (whether to pursue a univentricular or biventricular) be made at the initial operation, as attrition when the incorrect decision is made is high. There are several published criteria (Congenital Heart Surgeons’ Society critical stenosis calculator) to help surgeons decide which strategy to pursue.
Optimum strategy for repair of total anomalous pulmonary venous connection (TAPVC) remains a topic of some contention. Sutureless repair, formerly reserved for initial restenosis after conventional repair, has evolved in many centers to be the primary treatment of choice for high-risk patients. Defining whether sutureless repair should be considered in all patients with TAPVC will require further study.
A recent prospective, randomized, multi-institutional trial sponsored by the National Institutes of Health, the Systemic Ventricle Reconstruction (SVR) trial, compared the outcomes of neonates with hypoplastic left heart syndrome having either a modified Blalock-Taussig shunt versus a right ventricle-to-pulmonary artery (RV-PA) shunt. The SVR trial demonstrated that transplantation-free survival 12 months after randomization was higher with the RV-PA shunt than with the modified Blalock-Taussig shunt. However, data collected over a mean follow-up period of 32 ± 11 months showed a nonsignificant difference in transplantation-free survival between the two groups.
Outcomes have improved substantially over time in congenital heart surgery, and most complex lesions can be operated in early infancy. Neurologic protection, however, remains a key issue in the care of neonates undergoing surgery with cardiopulmonary bypass and deep hypothermic circulatory arrest. New monitoring devices and perioperative strategies are currently under investigation. Attention in the field has shifted currently from analyses of perioperative mortality, which for most lesions is under 10%, to longer-term outcomes, including quality of life and neurologic function.
INTRODUCTION
Congenital heart surgery is a dynamic and evolving field. The last 20 years have brought about rapid developments in technology, emphasis on a multidisciplinary approach to treatment, and a more thorough understanding of both the anatomy and pathophysiology of congenital heart disease, leading to the improved care of these challenging patients.
These advancements have created and sustained a paradigm shift in the field of congenital heart surgery. The traditional strategy of initial palliation followed by definitive correction at a later age, which had pervaded the thinking of most surgeons, began to evolve to one emphasizing early repair, even in the tiniest patients. Furthermore, some of the defects that were virtually uniformly fatal (such as hypoplastic left heart syndrome) are now successfully treated with aggressive forms of palliation using cardiopulmonary bypass, resulting in outstanding survival for many of these children.
Because the goal in most cases of congenital heart disease (CHD) is now early repair, as opposed to subdividing lesions into cyanotic or noncyanotic lesions, a more appropriate classification scheme divides particular defects into three categories based on the feasibility of achieving this goal: (a) defects that have no reasonable palliation and for which repair is the only option; (b) defects for which repair is not possible and for which palliation is the only option; and (c) defects that can either be repaired or palliated in infancy. It bears mentioning that all defects in the second category are those in which the appropriate anatomic components either are not present, as in hypoplastic left heart syndrome, or cannot be created from existing structures.
DEFECTS WHERE REPAIR IS THE ONLY OR BEST OPTION
An atrial septal defect (ASD) is defined as an opening in the interatrial septum that enables the mixing of blood from the systemic venous and pulmonary venous circulations.
The atrial and ventricular septa form between the third and sixth weeks of fetal development. After the paired heart tubes fuse into a single tube folded onto itself, the distal portion of the tube causes an indentation to form in the roof of the common atrium. Near this portion of the roof, the septum primum arises and extends into a crescentic formation toward the atrioventricular (AV) junction. The gap remaining between the septum primum and the developing tissues of the AV junction is called the ostium primum. Before the septum primum fuses completely with the endocardial cushions, a series of fenestrations appear in the septum primum that coalesce into the ostium secundum. During this coalescence, the septum secundum grows downward from the roof of the atrium, parallel to and to the right of the septum primum. The septum primum does not fuse, but creates an oblique pathway, called the foramen ovale, within the interatrial septum. After birth, the increase in left atrial pressure normally closes this pathway, obliterating the interatrial connection.
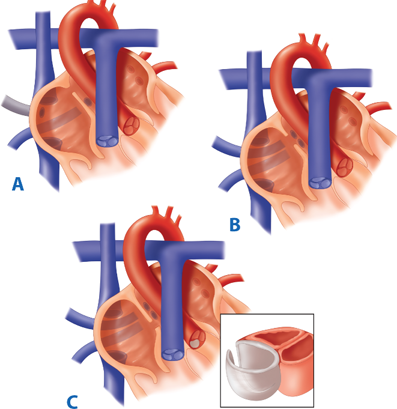
ASDs can be classified into three different types: (a) sinus venosus defects, comprising approximately 5% to 10% of all ASDs; (b) ostium primum defects, which are more correctly described as partial AV canal defects; and (c) ostium secundum defects, which are the most prevalent subtype, comprising 80% of all ASDs (Fig. 20-1).1
Figure 20-1.
The anatomy of atrial septal defects. In the sinus venosus type (A), the right upper and middle pulmonary veins frequently drain to the superior vena cava or right atrium. B. Secundum defects generally occur as isolated lesions. C. Primum defects are part of a more complex lesion and are best considered as incomplete atrioventricular septal defects. (Reproduced with permission from Greenfield LJ, Mulholland MW, Oldham KT, et al, eds. Surgery: Scientific Principles and Practice. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:1444.)
ASDs result in an increase in pulmonary blood flow secondary to left-to-right shunting through the defect. The direction of the intracardiac shunt is predominantly determined by the compliance of the respective ventricles. In utero, the distensibility, or compliance, of the right and left ventricles is equal, but postnatally the left ventricle (LV) becomes less compliant than the right ventricle (RV). This shift occurs because the resistance of the downstream vascular beds changes after birth. The pulmonary vascular resistance falls with the infant’s first breath, decreasing RV pressure, whereas the systemic vascular resistance rises dramatically, increasing LV pressure. The increased LV pressure creates a thicker muscle mass, which offers a greater resistance to diastolic filling than does the RV; thus, the majority of flow through the ASD occurs from left to right. The greater volume of blood returning to the right atrium causes volume overload in the RV, but because of its lower muscle mass and low-resistance output, it easily distends to accommodate this load.
The long-term consequences of RV volume overload include hypertrophy with elevated RV end-diastolic pressure and a relative pulmonary stenosis across the pulmonary valve, because it cannot accommodate the increased RV flow. The resistance at the level of the pulmonary valve then contributes a further pressure load on the RV, which accelerates RV hypertrophy. Compliance gradually decreases as the right ventricular pressure approaches systemic pressure, and the size of the left-to-right shunt decreases. Patients at this stage have a balanced circulation and may deceptively appear less symptomatic.
A minority of patients with ASDs develop progressive pulmonary vascular changes as a result of chronic overcirculation. The increased pulmonary vascular resistance in these patients leads to an equalization of left and right ventricular pressures, and their ratio of pulmonary (Qp) to systemic flow (Qs), Qp:Qs, will approach 1.2 This does not mean, however, that there is no intracardiac shunting, only that the ratio between the left-to-right component and the right-to-left component is equal.
The ability of the RV to recover normal function is related to the duration of chronic overload, because those undergoing ASD closure before age 10 years have a better likelihood of achieving normal RV function in the postoperative period.3
The physiology of sinus venosus ASDs is similar to that discussed earlier, except that these are frequently accompanied by anomalous pulmonary venous drainage. This often results in significant hemodynamic derangements that accelerate the clinical course of these infants.
The same increase in symptoms is true for those with ostium primum defects because the associated mitral insufficiency from the “cleft” mitral valve can lead to more atrial volume load and increased atrial level shunting.
Patients with ASDs may present with few physical findings. Auscultation may reveal prominence of the first heart sound with fixed splitting of the second heart sound. This results from the relatively fixed left-to-right shunt throughout all phases of the cardiac cycle. A diastolic flow murmur indicating increased flow across the tricuspid valve may be discerned, and frequently, an ejection flow murmur can be heard across the pulmonary valve. A right ventricular heave and increased intensity of the pulmonary component of the second heart sound indicates pulmonary hypertension and possible unrepairability.
Chest radiographs in the patient with an ASD may show evidence of increased pulmonary vascularity, with prominent hilar markings and cardiomegaly. The electrocardiogram shows right axis deviation with an incomplete bundle-branch block. When right bundle-branch block is associated with a leftward or superior axis, an AV canal defect should be strongly suspected.
Diagnosis is clarified by two-dimensional echocardiography, and use of color-flow mapping facilitates an understanding of the physiologic derangements created by the defects. Older children and adults with unrepaired ASDs may present with stroke or systemic embolism from paradoxical embolism or atrial arrhythmias from dilation of the right atrium.
Echocardiography also enables the clinician to estimate the amount of intracardiac shunting, can demonstrate the degree of mitral regurgitation in patients with ostium primum defects, and with the addition of microcavitation, can assist in the detection of sinus venosus defects.
The advent of two-dimensional echocardiography with color-flow Doppler has largely obviated the need for cardiac catheterization because the exact nature of the ASD can be precisely defined by echocardiography alone. However, in cases where the patient is older than age 40 years, catheterization can quantify the degree of pulmonary hypertension present, because those with a fixed pulmonary vascular resistance greater than 12 U/mL are considered inoperable.4 Cardiac catheterization also can be useful in that it provides data that enable the calculation of Qp and Qs so that the magnitude of the intracardiac shunt can be determined. The ratio (Qp:Qs) can then be used to determine whether closure is indicated in equivocal cases, because a Qp:Qs greater than 1.5:1 is generally accepted as the threshold for surgical intervention. Finally, in patients older than age 40 years, cardiac catheterization can be important to disclose the presence of coronary artery disease.
In general, ASDs are closed when patients are between 4 and 5 years of age. Children of this size can usually be operated on without the use of blood transfusion and generally have excellent outcomes. Patients who are symptomatic may require repair earlier, even in infancy. Some surgeons, however, advocate routine repair in infants and children, as even smaller defects are associated with the risk of paradoxical embolism. In a recent review by Reddy and colleagues, 116 neonates weighing less than 2500 g who underwent repair of simple and complex cardiac defects with the use of cardiopulmonary bypass were found to have no intracerebral hemorrhages, no long-term neurologic sequelae, and a low operative mortality rate (10%). These results correlated with the length of cardiopulmonary bypass and the complexity of repair.5 These investigators also found an 80% actuarial survival at 1 year and, more importantly, that growth following complete repair was equivalent to weight-matched neonates free from cardiac defects.5
ASDs can be repaired in a facile manner using standard cardiopulmonary bypass (CPB) techniques through a midline sternotomy approach. The details of the repair itself are generally straightforward. An oblique atriotomy is made, the position of the coronary sinus and all systemic and pulmonary veins are determined, and the rim of the defect is completely visualized. Closure of ostium secundum defects is accomplished either by primary repair or by insertion of a patch that is sutured to the rim of the defect. The decision of whether patch closure is necessary can be determined by the size and shape of the defect as well as by the quality of the edges.
The type of repair used for sinus venosus ASDs associated with partial anomalous pulmonary venous connection is dictated by the location of the anomalous pulmonary veins. If the anomalous veins connect to the atria or to the superior vena cava caudal to where the cava is crossed by the right pulmonary artery, the ASD can be repaired by inserting a patch, with redirection of the pulmonary veins behind the patch to the left atrium. Care must be taken with this approach to avoid obstruction of the pulmonary veins or the superior vena cava, although usually the superior vena cava is dilated and provides ample room for patch insertion. If the anomalous vein connects to the superior vena cava cranial to the right pulmonary artery, an alternative technique, the Warden procedure, may be necessary. In this operation, the superior vena cava is transected cranial to the connection of the anomalous vein (usually the right superior pulmonary vein). The caudal end of the transected cava is oversewn. The cranial end of the transected cava is anastomosed to the auricle of the right atrium. Inside the atrium, a patch is used to redirect pulmonary venous blood flow to the left atrium. In contrast to the repair for a defect where the pulmonary veins enter the right atrium or the superior vena cava below the right pulmonary artery, the patch covers the superior vena caval right atrial junction so that blood from the anomalous pulmonary vein that enters the cava is directed to the left atrium. Blood returning from the upper body enters the right atrium via the anastomosis between the superior vena cava and the right atrial appendage.
Traditional operative strategies, such as pericardial or synthetic patch closure, have been well established, with a low complication rate and a mortality rate of zero among patients without pulmonary hypertension.6 The most frequently reported immediate complications include postpericardiotomy syndrome and atrial arrhythmias. Beyond immediate postoperative outcomes, long-term outcomes following surgical closure (up to 20 years) document the low attrition rate and durability of functional status benefit. Importantly, however, atrial arrhythmias are not completely mitigated by closure and can occur in 10% to 40% of patients, especially in older patients (>40 years) or those with pre-existing arrhythmias.7 Kutty and colleagues8 followed 300 patients from their institution, 152 of whom had surgical closure. Late mortality at 10 years was 3%, and functional health status had declined in only 15 patients during follow-up. Recently, there have been an increasing number of reports regarding the results following surgical closure among elderly patients (>60 years of age), which demonstrate equivalent survival to younger patients, albeit with slightly higher complication rates.8,9,10 Hanninen and colleagues11 studied 68 patients between 68 and 86 years at their institution undergoing either surgical (n = 13) or device (n = 54) closure. Although the 23% incidence of major complications (including pneumothorax, heart failure, and pneumonia) was higher than that recently reported by Mascio et al12 using the Society of Thoracic Surgeons’ Congenital Database (20%) or a single-institution review by Hopkins et al13 (12%), there were no operative deaths among the elderly cohort. Moreover, after ASD closure, echocardiographic indices of right ventricular size and function were significantly improved from preoperative values, and functional capacity as measured by standardized survey instruments was also significantly improved.
Because of the uniformly excellent outcomes with traditional surgery, attention has shifted to improving the cosmetic result and minimizing hospital stay and convalescence. Multiple strategies have been described to achieve these aims, including the right submammary incision with anterior thoracotomy, limited bilateral submammary incision with partial sternal split, and limited midline incision with partial sternal split. Some surgeons use either video-assisted thoracic surgery (VATS) in conjunction with the submammary and transxiphoid approaches to facilitate closure within a constricted operative field or totally endoscopic repair in selected patients.14,15,16 Use of robotics has also been reported in a small series of 12 adult patients by Argenziano and colleagues.15 The morbidity and mortality of all of these approaches are comparable to those of the traditional median sternotomy; however, each has technical drawbacks. Operative precision must be maintained with limited exposure in any minimally invasive technique. Extended CPB and aortic cross-clamp times, coupled with increased cost, may limit the utility of totally endoscopic or robotic-assisted ASD closure except at limited centers. Certain approaches have a specific patient population in whom they are applicable. For example, the anterolateral thoracotomy should not be employed in prepubescent girls because it will interfere with breast development. Most totally endoscopic approaches are not feasible in very young patients due to the size of the thoracoscopic ports. Despite these potential drawbacks, however, in carefully selected patients, minimally invasive techniques have demonstrated benefits. Luo and associates performed a prospective randomized study comparing ministernotomy (division of the upper sternum for aortic and pulmonary lesions and the lower sternum for septal lesions) to full sternotomy in 100 consecutive patients undergoing repair of septal lesions.16 The patients in the ministernotomy group had longer procedure times (by 15 to 20 minutes), but had less bleeding and shorter hospital stays. Consistent with these initiatives, conversion of “low-risk” patients undergoing minimally invasive ASD closure to an ambulatory population (discharge from hospital within 24 hours) has recently been described.17
First performed in 1976, transcatheter closure of ASDs with the use of various occlusion devices is gaining widespread acceptance.18 Certain types of ASDs, including patent foramen ovale, secundum defects, and some fenestrated secundum defects, are amenable to device closure, as long as particular anatomic criteria (e.g., an adequate superior and inferior rim for device seating and distance from the AV valve) are met. Since the introduction of percutaneous closure, there has been a dramatic rise in device closure prevalence to the point where device closure has supplanted surgical therapy as the dominant treatment modality for secundum ASD.19 A study from Karamlou et al19 recently found that ASD and patent foramen ovale closures per capita increased dramatically from 1.08 per 100,000 population in 1988 to 2.59 per 100,000 population in 2005, an increase of 139%. When analyzed by closure type, surgical closure increased by only 24% (from 0.86 per 100,000 population in 1988 to 1.07 per 100,000 in 2005), whereas transcatheter closure increased by 3475% (from 0.04 per 100,000 population in 1988 to 1.43 per 100,000 in 2005). Importantly, this study determined that the paradigm shift favoring transcatheter closure has occurred mainly due to increased prevalence of closure in adults over age 40 years rather than an increase in closure in infants or children.
Despite the simplicity of ASD repair, there are a myriad of options for patients and physicians who care for patients with CHD. The patient population that might benefit from closure (whether device or surgical) is likely to increase, challenging current ideas and treatment algorithms that optimize outcomes.
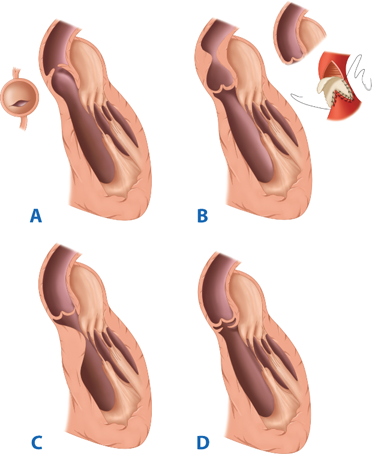
The spectrum of aortic valve abnormality represents the most common form of CHD, with the great majority of patients being asymptomatic until midlife. Obstruction of the left ventricular outflow tract (LVOT) occurs at multiple levels: subvalvular, valvular, and supravalvular (Fig. 20-2). The critically stenotic aortic valve in the neonate or infant is commonly unicommissural or bicommissural, with thickened, dysmorphic, and myxomatous leaflet tissue and a reduced cross-sectional area at the valve level. Associated left-sided lesions are often present. In a review of 32 cases from the Children’s Hospital in Boston, 59% had unicommissural valves and 40% had bicommissural valves.20 Associated lesions were frequent, occurring in 88% of patients, most commonly patent ductus arteriosus, mitral regurgitation, and hypoplastic LV. Endocardial fibroelastosis also is common among infants with critical aortic stenosis (AS). In this condition, the LV is largely nonfunctional, and these patients are not candidates for balloon valvotomy, simple valve replacement, or repair, because the LV is incapable of supporting the systemic circulation. Often, the LV is markedly hypertrophic with a reduced cavity size, but on rare occasion, a dilated LV, reminiscent of overt heart failure, is encountered.20
Figure 20-2.
The anatomy of the types of congenital aortic stenosis. A. Valvar aortic stenosis. B. Supravalvar aortic stenosis and its repair (insert). C. Tunnel-type subvalvar aortic stenosis. D. Membranous subvalvar aortic stenosis. (Reproduced with permission from Greenfield LJ, Mulholland MW, Oldham KT, et al, eds. Surgery: Scientific Principles and Practice. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001:1448.)
The unique intracardiac and extracardiac shunts present in fetal life allow even neonates with critical AS to survive. In utero, left ventricular hypertrophy and ischemia cause left atrial hypertension, which reduces the right-to-left flow across the foramen ovale. In severe cases, a reversal of flow may occur, causing right ventricular volume loading. The RV then provides the entire systemic output via the patent ductus arteriosus (ductal-dependent systemic blood flow). Although cardiac output is maintained, the LV suffers continued damage as the intracavitary pressure precludes adequate coronary perfusion, resulting in LV infarction and subendocardial fibroelastosis. The presentation of the neonate with critical AS is then determined by the morphology of the LV and other left-sided heart structures, the degree of left ventricular dysfunction, and the completeness of the transition from a parallel circulation to an in-series circulation (i.e., on closure of the foramen ovale and the ductus arteriosus). Those infants with mild-to-moderate AS in whom LV function is preserved are asymptomatic at birth. The only abnormalities may be a systolic ejection murmur and electrocardiogram (ECG) evidence of left ventricular hypertrophy. However, those neonates with severe AS and compromised LV function are unable to provide adequate cardiac output at birth and will present in circulatory collapse once the ductus closes, with dyspnea, tachypnea, irritability, narrowed pulse pressure, oliguria, and profound metabolic acidosis.7,21 If ductal patency is maintained, systemic perfusion will be provided by the RV via ductal flow, and cyanosis may be the only finding.
Neonates and infants with severe valvular AS may have a relatively nonspecific history of irritability and failure to thrive. Angina, if present, is usually manifested by episodic, inconsolable crying that coincides with feeding. As discussed previously, evidence of poor peripheral perfusion, such as extreme pallor, indicates severe LVOT obstruction. Differential cyanosis is an uncommon finding, but is present when enough antegrade flow occurs only to maintain normal upper body perfusion, while a large patent ductus arteriosus produces blue discoloration of the abdomen and legs.
Physical findings include a systolic ejection murmur, although a quiet murmur may paradoxically indicate a more severe condition with reduced cardiac output. A systolic click correlates with a valvular etiology of obstruction. As LV dysfunction progresses, evidence of congestive heart failure occurs.
The chest radiograph is variable but may show dilatation of the aortic root, and the ECG often demonstrates LV hypertrophy. Echocardiography with Doppler flow is extremely useful in establishing the diagnosis, as well as quantifying the transvalvular gradient.22 Furthermore, echocardiography can facilitate evaluation for the several associated defects that can be present in critical neonatal AS, including mitral stenosis, LV hypoplasia, LV endocardial fibroelastosis, subaortic stenosis, VSD, or coarctation. The presence of any or several of these defects has important implications related to treatment options for these patients. Although cardiac catheterization is not routinely performed for diagnostic purposes, it can be invaluable as part of the treatment algorithm if the lesion is amenable to balloon valvotomy.
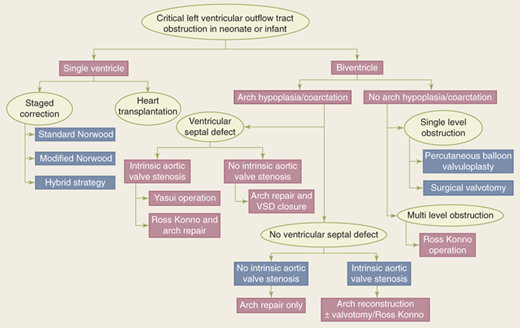
The first decision that must be made in the neonate with critical LVOT obstruction is whether the patient is a candidate for biventricular or univentricular repair. Central to this decision is assessment of the degree of hypoplasia of the LV and other left-sided structures. Alsoufi and colleagues23 recently described a rational approach to the neonate with critical LVOT obstruction (Fig. 20-3). The infant with severe AS requires urgent intervention. Preoperative stabilization, however, has dramatically altered the clinical algorithm and outcomes for this patient population.19,21 The preoperative strategy begins with endotracheal intubation and inotropic support. Prostaglandin infusion is initiated to maintain ductal patency, and confirmatory studies are performed prior to operative intervention.
Figure 20-3.
Treatment algorithm for neonates and infants with critical left ventricular outflow tract obstruction. Patients can be initially triaged to either a single or a biventricular approach depending on presenting morphologic, demographic, and institutional factors. VSD = ventricular septal defect. (From Alsoufi B, et al. Management options in neonates and infants with critical left ventricular outflow tract obstruction. Eur J Cardiothorac Surg. 2007;31:1013. Fig 1. By permission of Oxford University Press.)
Therapy is generally indicated in the presence of a transvalvular gradient of 50 mmHg with associated symptoms including syncope, CHF, or angina, or if a gradient of 50 to 75 mmHg exists with concomitant ECG evidence of LV strain or ischemia. In the critically ill neonate, there may be little gradient across the aortic valve because of poor LV function. These patients depend on patency of the ductus arteriosus to provide systemic perfusion from the RV, and all ductal-dependent patients with critical AS require treatment. However, the decision regarding treatment options must be based on a complete understanding of associated defects. For example, in the presence of a hypoplastic LV (left ventricular end-diastolic volume <20 mL/m2) or a markedly abnormal mitral valve, isolated aortic valvotomy should not be performed because studies have demonstrated high mortality in this population following isolated valvotomy.24
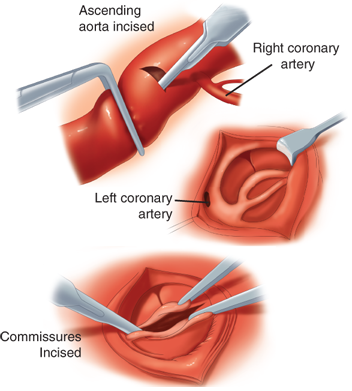
Patients who have an LV capable of providing systemic output are candidates for intervention to relieve AS, generally through balloon valvotomy. Very rarely, if catheter-based therapy is not an option, relief of valvular AS in infants and children can be accomplished with surgical valvotomy using standard techniques of CPB and direct exposure to the aortic valve. A transverse incision is made in the ascending aorta above the sinus of Valsalva, extending close to, but not into, the noncoronary sinus. Exposure is attained with placement of a retractor into the right coronary sinus. After inspection of the valve, the chosen commissure is incised to within 1 to 2 mm of the aortic wall (Fig. 20-4).
Figure 20-4.
Aortic valvotomy with cardiopulmonary bypass. A transverse incision is made in the ascending aorta above the sinuses of Valsalva, extending close to, but not into, the noncoronary sinus. Exposure is accomplished with placement of a retractor into the right coronary sinus. After inspection of the valve, the chosen commissure is incised to within 1 to 2 mm of the aortic wall. (Reproduced with permission from Doty DB. Cardiac Surgery: A Looseleaf Workbook and Update Service. Chicago: Year Book; 1986. Copyright Elsevier)
Balloon valvotomy performed in the catheterization lab is the procedure of choice for reduction of transvalvular gradients in symptomatic infants and children. This procedure is an ideal palliative option because mortality from surgical valvotomy can be high due to the critical nature of these patients’ condition. Furthermore, balloon valvotomy provides relief of the valvular gradient and allows future surgical intervention (which is generally required in most patients when a larger prosthesis can be implanted) to be performed on an unscarred chest. An important issue when planning aortic valvotomy, whether percutaneously or via open surgical technique, is the risk of inducing hemodynamically significant aortic regurgitation. Induction of more than moderate aortic regurgitation is poorly tolerated in the infant with critical AS and may require an urgent procedure to replace or repair the aortic valve.
In general, catheter-based balloon valvotomy has supplanted open surgical valvotomy. The decision regarding the most appropriate method to use depends on several factors, including the available medical expertise, the patient’s overall status and hemodynamics, and the presence of associated cardiac defects requiring repair.25 Although evidence is emerging to the contrary, simple valvotomy, whether performed using percutaneous or open technique, is generally considered a palliative procedure. The goal is to relieve LVOT obstruction without producing clinically significant regurgitation, in order to allow sufficient annular growth for eventual aortic valve replacement. The majority of infants who undergo aortic valvotomy will require further intervention on the aortic valve within 10 years following initial intervention.26
Valvotomy may result in aortic insufficiency that, while not important enough to require intervention in infancy, may alone or in combination with AS result in the need for an aortic valve replacement. Neonates with severely hypoplastic LVs or significant LV endocardial fibroelastosis may not be candidates for two-ventricle repair and are treated the same as infants with the hypoplastic left heart syndrome (HLHS), which is discussed later (see later section, Hypoplastic Left Heart Syndrome).
Many surgeons previously avoided aortic valve replacement for AS in early childhood because the more commonly used mechanical valves would be outgrown and require replacement later and the obligatory anticoagulation for mechanical valves resulted in a substantial risk for complications. In addition, mechanical valves had an important incidence of bacterial endocarditis or perivalvular leak requiring re-intervention.
The use of allografts and the advent of the Ross procedure have largely obviated these issues and made early definitive correction of critical AS a viable option.19,27,28 Donald Ross first described transposition of the pulmonary valve into the aortic position with allograft reconstruction of the pulmonary outflow tract in 1967.27 The result of this operation is a normal trileaflet semilunar valve made of a patient’s native tissue with the potential for growth to adult size in the aortic position in place of the damaged aortic valve (Fig. 20-5). The Ross procedure has become a useful option for aortic valve replacement in children, because it has improved durability and can be performed with acceptable morbidity and mortality rates. The placement of a pulmonary conduit, which does not grow and becomes calcified and stenotic over time, does obligate the patient to reoperation to replace the RV-to-pulmonary artery conduit. Karamlou and colleagues29 recently reviewed the outcomes and associated risk factors for aortic valve replacement in 160 children from the Hospital for Sick Children in Toronto. They found that younger age, lower operative weight, concomitant performance of aortic root replacement or reconstruction, and use of prosthesis type other than a pulmonary autograft were significant predictors of death, whereas the use of a bioprosthetic or allograft valve type and earlier year of operation were identified as significant risk factors for repeated aortic valve replacement. Autograft use was associated with a blunted progression of the peak prosthetic valve gradient and a rapid decrease in the left ventricular end-diastolic dimension (Fig. 20-6). In agreement with these findings, Lupinetti and Jones28 compared allograft aortic valve replacement with the Ross procedure and found a more significant transvalvular gradient reduction and regression of left ventricular hypertrophy in those patients who underwent the Ross procedure. In some cases, the pulmonary valve may not be usable because of associated defects or congenital absence. These children are not candidates for the Ross procedure and are now most frequently treated with cryopreserved allografts (cadaveric human aortic valves). At times, there may be a size discrepancy between the right ventricular outflow tract (RVOT) and the LVOT, especially in cases of severe critical AS in infancy. For these cases, the pulmonary autograft is placed in a manner that also provides enlargement of the aortic annulus (Ross/Konno) (Fig. 20-7).
Figure 20-5.
A through C. The pulmonary autograft for aortic valve replacement. The aortic valve and adjacent aorta are excised, preserving buttons of aortic tissue around the coronary ostia. The pulmonary valve and main pulmonary artery are excised and transferred to the aortic position. The coronary buttons are then attached to the neoaortic root. A pulmonary allograft is inserted to re-establish the right ventricular outflow tract. (From Kouchokos NT, Davila-Roman VG, Spray TL, et al. Replacement of the aortic root with a pulmonary autograft in children and young adults with aortic-valve disease. N Engl J Med. 1994;330:1.)
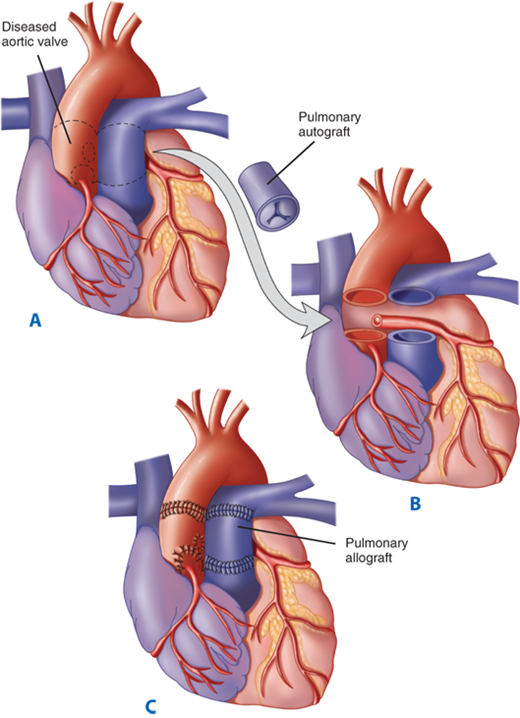
Figure 20-6.
Predicted progression of the peak instantaneous prosthetic valve gradient after initial aortic valve replacement (AVR) stratified by prosthesis type (autograft vs. nonautograft) for a hypothetical patient of 3 years undergoing AVR in 1990. Solid lines represent point estimates from a mixed linear regression model (surrounded by their 90% confidence intervals in dashed lines). AoV = aortic valve. (Reproduced with permission from Karamlou T, et al. Outcomes and associated risk factors for aortic valve replacement in 160 children: A competing risks analysis. Circulation. 2005;112:3462.)

Figure 20-7.
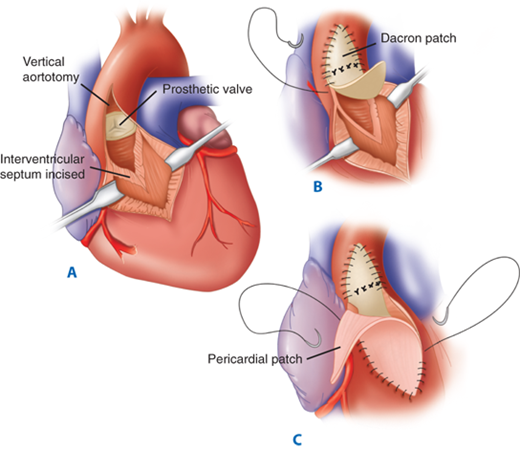
The Konno-Rastan aortoventriculoplasty permits significant enlargement of the aortic annulus and subaortic region. A. A vertical aortotomy is made to the left of the right coronary artery and extended into the right ventricular outflow tract. After excising the aortic valve, the interventricular septum is incised and an aortic valve prosthesis is secured within the enlarged annulus. B. A Dacron patch that is attached to the sewing ring of the prosthesis closes the interventricular septum and the aortotomy. C. A separate pericardial patch closes the right ventriculotomy. (Reproduced with permission from Misbach GA, Turley K, Ullyot DJ, et al. Left ventricular outflow enlargement by the Konno procedure. J Thorac Cardiovasc Surg. 1982;84:696. Copyright Elsevier.)
Subvalvular AS occurs beneath the aortic valve and may be classified as discrete or tunnel-like (diffuse). A thin, fibromuscular diaphragm immediately proximal to the aortic valve characterizes discrete subaortic stenosis. This diaphragm typically extends for 180° or more in a crescentic or circular fashion, often attaching to the mitral valve as well as the interventricular septum.21 The aortic valve itself is usually normal in this condition, although the turbulence imparted by the subvalvular stenosis may affect leaflet morphology and valve competence.
Diffuse subvalvular AS results in a long, tunnel-like obstruction that may extend to the left ventricular apex. In some individuals, there may be difficulty in distinguishing between hypertrophic cardiomyopathy and diffuse subaortic stenosis. Operation for subvalvular AS is indicated with a gradient exceeding 30 mmHg, in the presence of aortic valve insufficiency, or when symptoms indicating LVOT obstruction are present.30 Given that repair of isolated discrete subaortic stenosis can be done with low rates of morbidity and mortality, some surgeons advocate repair in all cases of discrete AS to avoid progression of the stenosis and the development of aortic insufficiency, although more recent data demonstrates that subaortic resection should be delayed until the LV gradient exceeds 30 mmHg because most children with an initial LV gradient less than 30 mmHg have quiescent disease.31 Diffuse AS is a more complex lesion and often requires aortoventriculoplasty as previously described. Results are generally excellent, with operative mortality less than 5%.32
Supravalvular AS occurs more rarely and also can be classified into a discrete type, which produces an hourglass deformity of the aorta, and a diffuse form that can involve the entire arch and brachiocephalic arteries. The aortic valve leaflets are usually normal, but in some cases, the leaflets may adhere to the supravalvular stenosis, thereby narrowing the sinuses of Valsalva in diastole and restricting coronary artery perfusion. In addition, accelerated intimal hyperplastic changes in the coronary arteries can be demonstrated in these patients because the proximal position of the coronary arteries subjects them to abnormally high perfusion pressures.
The signs and symptoms of supravalvular AS are similar to other forms of LVOT obstruction. An asymptomatic murmur is the presenting manifestation in approximately half of these patients. Syncope, poor exercise tolerance, and angina may all occur with nearly equal frequency. Supravalvar AS is associated with Williams’ syndrome, a constellation of elfin facies, mental retardation, and hypercalcemia.33 Following routine evaluation, cardiac catheterization should be performed in order to delineate coronary anatomy, as well as to delineate the degree of obstruction. A gradient of 50 mmHg or greater is an indication for operation. However, the clinician must be cognizant of any coexistent lesions, most commonly pulmonic stenosis, which may add complexity to the repair.
The localized form of supravalvular AS is treated by creating an inverted Y-shaped aortotomy across the area of stenosis, straddling the right coronary artery. The obstructing shelf is then excised and a pantaloon-shaped patch is used to close the incision.21
The diffuse form of supravalvular stenosis is more variable, and the particular operative approach must be tailored to each specific patient’s anatomy. In general, either an aortic endarterectomy with patch augmentation can be performed, or if the narrowing extends past the aorta arch, a prosthetic graft can be placed between the ascending and descending aorta. Operative results for discrete supravalvular AS are generally good, with a hospital mortality of less than 1% and an actuarial survival rate exceeding 90% at 20 years.34 In contrast, however, the diffuse form is more hazardous to repair and carried a mortality of 15% in a recent series.34,35
The ductus arteriosus is derived from the sixth aortic arch and normally extends from the main or left pulmonary artery to the upper descending thoracic aorta, distal to the left subclavian artery. In the normal fetal cardiovascular system, ductal flow is considerable (approximately 60% of the combined ventricular output) and is directed exclusively from the pulmonary artery to the aorta.36 In infancy, the length of the ductus may vary from 2 to 8 mm, with a diameter of 4 to 12 mm.
Locally produced and circulating prostaglandin E2 (PGE2) and prostaglandin I2 (PGI2) induce active relaxation of the ductal musculature, maintaining maximal patency during the fetal period.37 At birth, increased pulmonary blood flow metabolizes these prostaglandin products, and absence of the placenta removes an important source of them, resulting in a marked decrease in these ductal-relaxing substances. In addition, release of histamines, catecholamines, bradykinin, and acetylcholine all promote ductal contraction. Despite all of these complex interactions, the rising oxygen tension in the fetal blood is the main stimulus causing smooth muscle contraction and ductal closure within 10 to 15 hours postnatally.38 Anatomic closure by fibrosis produces the ligamentum arteriosum connecting the pulmonary artery to the aorta.
Delayed closure of the ductus is termed prolonged patency, whereas failure of closure causes persistent patency, which may occur as an isolated lesion or in association with more complex congenital heart defects. In many of these infants with more complex congenital heart defects, either pulmonary or systemic perfusion may depend on ductal flow, and these infants may decompensate if exogenous PGE is not administered to maintain ductal patency.
The incidence of patent ductus arteriosus (PDA) is approximately 1 in every 2000 births; however, it increases dramatically with increasing prematurity.39 In some series, PDAs have been noted in 75% of infants of 28 to 30 weeks gestation. Persistent patency occurs more commonly in females, with a 2:1 ratio.39
PDA is not a benign entity, although prolonged survival has been reported. The estimated death rate for infants with isolated, untreated PDA is approximately 30%.40 The leading cause of death is congestive heart failure, with respiratory infection as a secondary cause. Endocarditis is more likely to occur with a small ductus and is rarely fatal if aggressive antibiotic therapy is initiated early.
After birth, in an otherwise normal cardiovascular system, a PDA results in a left-to-right shunt that depends on both the size of the ductal lumen and its total length. As the pulmonary vascular resistance falls 16 to 18 weeks postnatally, the shunt will increase, and its flow will ultimately be determined by the relative resistances of the pulmonary and systemic circulations.
The hemodynamic consequences of an unrestrictive ductal shunt are left ventricular volume overload with increased left atrial and pulmonary artery pressures, and right ventricular strain from the augmented afterload. These changes result in increased sympathetic discharge, tachycardia, tachypnea, and ventricular hypertrophy. The diastolic shunt results in lower aortic diastolic pressure and increases the potential for myocardial ischemia and underperfusion of other systemic organs, while the increased pulmonary flow leads to increased work of breathing and decreased gas exchange. Unrestrictive ductal flow may lead to pulmonary hypertension within the first year of life. These changes will be significantly attenuated if the size of the ductus is only moderate, and completely absent if the ductus is small.
Physical examination of the afflicted infant will reveal evidence of a hyperdynamic circulation with a widened pulse pressure and a hyperactive precordium. Auscultation demonstrates a systolic or continuous murmur, often termed a machinery murmur. Cyanosis is not present in uncomplicated isolated PDA.
The chest radiograph may reveal increased pulmonary vascularity or cardiomegaly, and the ECG may show LV strain, left atrial enlargement, and possibly RV hypertrophy. Echocardiogram with color mapping reliably demonstrates the patency of the ductus as well as estimates the shunt size. Cardiac catheterization is necessary only when pulmonary hypertension is suspected.
The presence of a persistent PDA is sufficient indication for closure because of the increased mortality and risk of endocarditis.2,4 In older patients with pulmonary hypertension, closure may not improve symptoms and is associated with much higher mortality.
In premature infants, aggressive intervention with indomethacin or ibuprofen to achieve early closure of the PDA is beneficial unless contraindications such as necrotizing enterocolitis or renal insufficiency are present.41 Term infants, however, are generally unresponsive to pharmacologic therapy with indomethacin, so mechanical closure must be undertaken once the diagnosis is established. This can be accomplished either surgically or with catheter-based therapy.12,42,43 Currently, transluminal placement of various occlusive devices, such as the Rashkind double-umbrella device or embolization with Gianturco coils, is in widespread use.42 However, there are a number of complications inherent with the use of percutaneous devices, such as thromboembolism, endocarditis, incomplete occlusion, vascular injury, and hemorrhage secondary to perforation.43 In addition, these techniques may not be applicable in very young infants, because the peripheral vessels do not provide adequate access for the delivery devices.
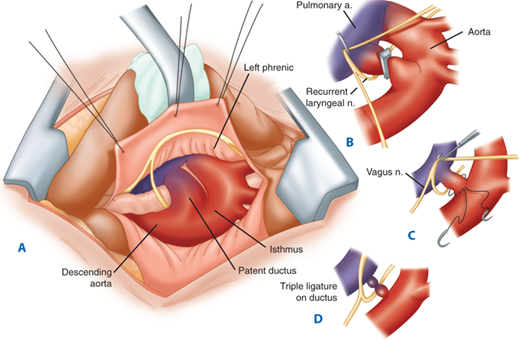
Surgical closure can be achieved via either open or video-assisted approaches. The open approach employs a posterior lateral thoracotomy in the fourth or fifth intercostal space on the side of the aorta (generally the left). The lung is then retracted anteriorly. In the neonate, the PDA is singly ligated with a surgical clip or permanent suture. In older patients, the PDA is triply ligated. Care must be taken to avoid the recurrent laryngeal nerve, which courses around the PDA. The PDA can also be ligated via a median sternotomy; however, this approach is generally reserved for patients who have additional cardiac or great vessel lesions requiring repair. Occasionally, a short, broad ductus, in which the dimension of its width approaches that of its length, will be encountered. In this case, division between vascular clamps with oversewing of both ends is advisable (Fig. 20-8). In extreme cases, the use of CPB to decompress the large ductus during ligation is an option.
Figure 20-8.
A. Surgeon’s perspective of infant patent ductus arteriosus exposed via a left thoracotomy. B. The pleura over the aortic isthmus is incised and mobilized. C and D. Technique of triple ligation. a. = artery; n. = nerve. (From Castaneda AR, Jonas RA, Mayer JE, et al. Cardiac Surgery of the Neonate and Infant. Philadelphia: W.B. Saunders; 1994:208, with permission. Copyright Elsevier.)
Video-assisted thoracoscopic occlusion, using metal clips, also has been described, although it offers few advantages over the standard surgical approach.12 Preterm newborns and children may do well with the thoracoscopic technique, while older patients (older than age 5 years) and those with smaller ducts (<3 mm) do well with coil occlusion. In fact, Moore and colleagues recently concluded from their series that coil occlusion is the procedure of choice for ducts smaller than 4 mm.44 Complete closure rates using catheter-based techniques have steadily improved. Comparative studies of cost and outcome between open surgery and transcatheter duct closure, however, have shown no overwhelming choice between the two modalities. Burke prospectively reviewed coil occlusion and VATS at Miami Children’s Hospital and found both options to be effective and less morbid than traditional thoracotomy.12
In premature infants, the surgical mortality is very low, although the overall hospital death rate is significant as a consequence of other complications of prematurity. In older infants and children, mortality is less than 1%. Bleeding, chylothorax, vocal cord paralysis, and the need for reoperation occur infrequently. With the advent of muscle-sparing thoracotomy, the risk of subsequent arm dysfunction or breast abnormalities is virtually eliminated.45
Coarctation of the aorta (COA) is defined as a luminal narrowing in the aorta that causes an obstruction to blood flow. This narrowing is most commonly located distal to the left subclavian artery. The embryologic origin of COA is a subject of some controversy. One theory holds that the obstructing shelf, which is largely composed of tissue found within the ductus, forms as the ductus involutes.46 The other theory holds that a diminished aortic isthmus develops secondary to decreased aortic flow in infants with enhanced ductal circulation.
Extensive collateral circulation develops, predominantly involving the intercostals and mammary arteries as a direct result of aortic flow obstruction. This translates into the well-known finding of “rib-notching” on chest radiograph, as well as a prominent pulsation underneath the ribs.
Other associated anomalies, such as ventricular septal defect, PDA, and ASD, may be seen with COA, but the most common is that of a bicuspid aortic valve, which can be demonstrated in 25% to 42% of cases.47
Infants with COA develop symptoms consistent with left ventricular outflow obstruction, including pulmonary overcirculation and, later, biventricular failure. In addition, proximal systemic hypertension develops as a result of mechanical obstruction to ventricular ejection, as well as hypoperfusion-induced activation of the renin-angiotensin-aldosterone system. Interestingly, hypertension is often persistent after surgical correction despite complete amelioration of the mechanical obstruction and pressure gradient.48 It has been shown that early surgical correction may prevent the development of long-term hypertension, which undoubtedly contributes to many of the adverse sequelae of COA, including the development of circle of Willis aneurysms, aortic dissection and rupture, and an increased incidence of coronary arteriopathy with resulting myocardial infarction.49
COA is likely to become symptomatic either in the newborn period if other anomalies are present or in the late adolescent period with the onset of left ventricular failure.
Physical examination will demonstrate a hyperdynamic precordium with a harsh murmur localized to the left chest and back. Femoral pulses will be dramatically decreased when compared to upper extremity pulses, and differential cyanosis may be apparent until ductal closure.
Echocardiography will reliably demonstrate the narrowed aortic segment, as well as define the pressure gradient across the stenotic segment. In addition, detailed information regarding other associated anomalies can be gleaned. Aortography is reserved for those cases in which the echocardiographic findings are equivocal.
The routine management of hemodynamically significant COA in all age groups has traditionally been surgical. Transcatheter repairs are used with increasing frequency in older patients and those with re-coarctation following surgical repair. Balloon dilatation of native coarctation in neonates has been recently utilized with poor results. The most common surgical techniques in current use are resection with end-to-end anastomosis or extended end-to-end anastomosis, taking care to remove all residual ductal tissue.50,51 Extended end-to-end anastomosis may also allow the surgeon to treat transverse arch hypoplasia, which is commonly encountered in infants with aortic coarctation.52,53 The subclavian flap aortoplasty is another repair, although it is used less frequently in the modern era because of the risk of late aneurysm formation and possible underdevlopment of the left upper extremity or ischemia.51 In this method, the left subclavian artery is transected and brought down over the coarcted segment as a vascularized patch. The main benefit of these techniques is that they do not involve the use of prosthetic materials, and evidence suggests that extended end-to-end anastomosis may promote arch growth, especially in infants with the smallest initial aortic arch diameters.52
Despite the benefits, however, extended end-to-end anastomosis may not be feasible when there is a long segment of coarctation or in the presence of previous surgery, because sufficient mobilization of the aorta above and below the lesion may not be possible. In this instance, prosthetic materials, such as a patch aortoplasty, in which a prosthetic patch is used to enlarge the coarcted segment, or an interposition tube graft must be employed.
The most common complications after COA repair are late restenosis and aneurysm formation at the repair site.54-56 Aneurysm formation is particularly common after patch aortoplasty when using Dacron material. In a large series of 891 patients, aneurysms occurred in 5.4% of the total, with 89% occurring in the group who received Dacron-patch aortoplasty and only 8% occurring in those who received resection with primary end-to-end anastomosis.54 A further complication, although uncommon, is lower-body paralysis resulting from ischemic spinal cord injury during the repair. This dreaded outcome complicates 0.5% of all surgical repairs, but its incidence can be lessened with the use of some form of distal perfusion, preferably left heart bypass with the use of femoral arterial or distal thoracic aorta for arterial inflow and the femoral vein or left atrium for venous return.50 These techniques are generally reserved for older patients with complex coarctations that may need prolonged aortic cross clamp times for repair, often in the setting of large collateral vessels and/or previous surgery.
Hypertension is also well recognized following repair of COA. Bouchart and colleagues reported that in a cohort of 35 hypertensive adults (mean age, 28 years) undergoing repair, despite a satisfactory anatomic outcome, only 23 patients were normotensive at a mean follow-up period of 165 months.55 Likewise, Bhat and associates reported that in a series of 84 patients (mean age at repair, 29 years), 31% remained hypertensive at a mean follow-up of 5 years following surgery.56
Although operative repair is still the gold standard, treatment of COA by catheter-based intervention has become more widespread. Both balloon dilatation and primary stent implantation have been used successfully. The most extensive study of the results of balloon angioplasty reported on 970 procedures: 422 native and 548 recurrent COAs. Mean gradient reduction was 74% ± 24% for native and 70% ± 31% for recurrent COA.57 This demonstrated that catheter-based therapy could produce equally effective results both in recurrent and in primary COA, a finding with far-reaching implications in the new paradigm of multidisciplinary treatment algorithms for CHD. In the Valvuloplasty and Angioplasty of Congenital Anomalies (VACA) report, higher preangioplasty gradient, earlier procedure date, older patient age, and the presence of recurrent COA were independent risk factors for suboptimal procedural outcome.57
The gradient after balloon dilatation in most series is generally acceptable. However, there is a significant minority of patients (0%–26%) for whom the procedural outcome is suboptimal, with a postprocedure gradient of 20 mmHg or greater. These patients may be ideal candidates for primary stent placement. Restenosis is much less common in children, presumably reflecting the influence of vessel wall scarring and growth in the pediatric age group.
Deaths from the procedure also are infrequent (<1% of cases), and the main major complication is aneurysm formation, which occurs in 7% of patients.50 With stent implantation, many authors have demonstrated improved resolution of stenosis compared with balloon dilatation alone, yet the long-term complications on vessel wall compliance remain largely unknown because only mid-term data are widely available.
In summary, children younger than age 6 months with native COA should be treated with surgical repair, while those requiring intervention at later ages may be ideal candidates for balloon dilatation or primary stent implantation.50 Additionally, catheter-based therapy should be employed for those cases of restenosis following either surgical or primary endovascular management.
Truncus arteriosus is a rare anomaly, comprising between 1% and 4% of all cases of CHD.58 It is characterized by a single great artery that arises from the heart, overrides the ventricular septum, and supplies the pulmonary, systemic, and coronary circulations.
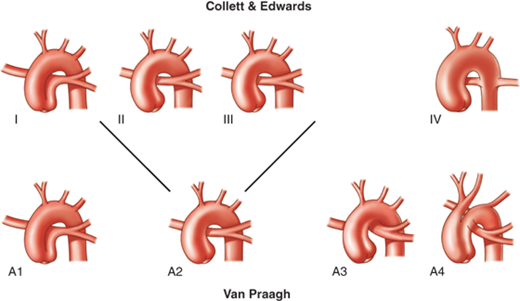
The two major classification systems are those of Collett and Edwards, described in 1949, and Van Praagh and Van Praagh, described in 1965 (Fig. 20-9).59,60 The Collett and Edwards classification focuses mainly on the origin of the pulmonary arteries from the common arterial trunk, whereas the Van Praagh system is based on the presence or absence of a VSD, the degree of formation of the aorticopulmonary septum, and the status of the aortic arch.
Figure 20-9.
There are similarities between the Collett and Edwards and the Van Praagh classifications of truncus arteriosus. Type I is the same as A1. Types II and III are grouped as a single type A2 because they are not significantly distinct embryologically or therapeutically. Type A3 denotes unilateral pulmonary artery with collateral supply to the contralateral lung (hemitruncus). Type A4 is truncus associated with interrupted aortic arch (13% of all cases of truncus arteriosus). (Reproduced with permission from Fyler DC. Truncus arteriosus. In: Fyler DC, ed. Nadas’ Pediatric Cardiology. Philadelphia: Hanley & Belfus; 1992:676. Copyright Elsevier. As adapted with permission from Hernanz-Schulman M, Fellows KE. Persistent truncus arteriosus: pathologic, diagnostic and therapeutic considerations. Semin Roentgenol. 1985;20:121. Copyright Elsevier.)
During embryonic life, the truncus arteriosus normally begins to separate and spiral into a distinguishable anterior pulmonary artery and posterior aorta. Persistent truncus, therefore, represents an arrest in embryologic development at this stage.61 Other implicated events include twisting of the dividing truncus because of ventricular looping, subinfundibular atresia, and abnormal location of the semilunar valve anlages.62
The neural crest may also play a crucial role in the normal formation of the great vessels, as experimental studies in chick embryos have shown that ablation of the neural crest results in persistent truncus arteriosus.63 The neural crest also develops into the pharyngeal pouches that give rise to the thymus and parathyroids, which likely explains the prevalent association of truncus arteriosus and DiGeorge’s syndrome.64
The annulus of the truncal valve usually straddles the ventricular septum in a “balanced” fashion; however, it is not unusual for it to be positioned predominantly over the RV, which increases the potential for LVOT obstruction following surgical repair. In the great majority of cases, the leaflets are thickened and deformed, which leads to valvular insufficiency. There are usually three leaflets (60%), but occasionally a bicuspid (50%) or even a quadricuspid valve (25%) is present.65
In truncus arteriosus, the pulmonary trunk bifurcates, with the left and right pulmonary arteries forming posteriorly and to the left in most cases. The caliber of the pulmonary arterial branches is usually normal, with stenosis or diffuse hypoplasia occurring in rare instances.
The coronary arteries may be normal; however, anomalies are not unusual and occur in 50% of cases.65,66 Many of these are relatively minor, although two variations are of particular importance, because they have implications in the conduct of operative repair. The first is that the left coronary ostium may arise high in the sinus of Valsalva or even from the truncal tissue at the margin of the pulmonary artery tissue. This coronary artery can be injured during repair when the pulmonary arteries are removed from the trunk or when the resulting truncal defect is closed. The second is that the right coronary artery can give rise to an important accessory anterior descending artery, which often passes across the RV in the exact location where the right ventriculotomy is commonly performed during repair.65,66
The main pathophysiologic consequences of truncus arteriosus are (a) the obligatory mixing of systemic and pulmonary venous blood at the level of the ventricular septal defect (VSD) and truncal valve, which leads to arterial saturations near 85%, and (b) the presence of a nonrestrictive left-to-right shunt, which occurs during both systole and diastole, the volume of which is determined by the relative resistances of the pulmonary and systemic circulations.65 Additionally, truncal valve stenosis or regurgitation, the presence of important LVOT obstruction, and stenosis of pulmonary artery branches can further contribute to both pressure- and volume-loading of the ventricles. The presence of these lesions often results in severe heart failure and cardiovascular instability early in life. Pulmonary vascular resistance may develop as early as 6 months of age, leading to poor results with late surgical correction.
Patients with truncus arteriosus usually present in the neonatal period, with signs and symptoms of congestive heart failure and mild to moderate cyanosis. A pansystolic murmur may be noted at the left sternal border, and occasionally a diastolic murmur may be heard in the presence of truncal regurgitation.
Chest radiography will be consistent with pulmonary overcirculation, and a right aortic arch can be appreciated 35% of the time. The ECG is usually nonspecific, demonstrating normal sinus rhythm with biventricular hypertrophy.
Echocardiography with Doppler color-flow or pulsed Doppler is diagnostic and usually provides sufficient information to determine the type of truncus arteriosus, the origin of the coronary arteries and their proximity to the pulmonary trunk, the character of the truncal valves, and the extent of truncal insufficiency.65 Cardiac catheterization can be helpful in cases where pulmonary hypertension is suspected or to further delineate coronary artery anomalies prior to repair.
The presence of truncus is an indication for surgery. Repair should be undertaken in the neonatal period or as soon as the diagnosis is established. Eisenmenger’s physiology, which is found primarily in older children, is the only absolute contraindication to correction.
Truncus arteriosus was first managed with pulmonary artery banding as described by Armer and colleagues in 1961.67 However, this technique led to only marginal improvements in 1-year survival rates because ventricular failure inevitably occurred. In 1967, however, complete repair was accomplished by McGoon and his associates based on the experimental work of Rastelli, who introduced the idea that an extracardiac valved conduit could be used to restore ventricular-to-pulmonary artery continuity.68 Over the next 20 years, improved survival rates led to uniform adoption of complete repair even in the youngest and smallest infants.58,65,69
Surgical correction entails the use of CPB. Repair is completed by separation of the pulmonary arteries from the aorta, closure of the aortic defect (occasionally with a patch) to minimize coronary flow complications, placement of a valved cryopreserved allograft or jugular venous valved conduit (Contegra) to reconstruct the RVOT, and VSD closure. Important branch pulmonary arterial stenosis should be repaired at the time of complete repair and can usually be accomplished with longitudinal allograft patch arterioplasty. Severe truncal valve insufficiency occasionally requires truncal valve replacement, which can be accomplished with a cryopreserved allograft.70
The results of complete repair of truncus have steadily improved. Ebert reported a 91% survival rate in his series of 77 patients who were younger than 6 months of age; later reports by others confirmed these findings and demonstrated that excellent results could be achieved in even smaller infants with complex-associated defects.11,69
Newer extracardiac conduits also have been developed and used with success, which has widened the repertoire of the modern congenital heart surgeon and improved outcomes.71 Severe truncal regurgitation, interrupted aortic arch, coexistent coronary anomalies, chromosomal or genetic anomalies, and age younger than 100 days are risk factors associated with perioperative death and poor outcome.
Total anomalous pulmonary venous connection (TAPVC) occurs in 1% to 2% of all cardiac malformations and is characterized by abnormal drainage of the pulmonary veins into the right heart, whether through connections into the right atrium or into its tributaries.72 Accordingly, the only mechanism by which oxygenated blood can return to the left heart is through an ASD, which is almost uniformly present with TAPVC.
Unique to this lesion is the absence of a definitive form of palliation. Thus, TAPVC with concomitant obstruction represents one of the only true surgical emergencies across the entire spectrum of congenital heart surgery.
The lungs develop from an outpouching of the foregut, and their venous plexus arises as part of the splanchnic venous system. TAPVC arises when the pulmonary vein evagination from the posterior surface of the left atrium fails to fuse with the pulmonary venous plexus surrounding the lung buds. In place of the usual connection to the left atrium, at least one connection of the pulmonary plexus to the splanchnic plexus persists. Accordingly, the pulmonary veins drain to the heart through a systemic vein (Fig. 20-10).
Figure 20-10.
Total anomalous pulmonary venous connection results when the primordial pulmonary vein fails to unite with the plexus of veins that surround the lung buds and is derived from the splanchnic venous plexus, including the cardinal veins and umbilicovitelline veins. (From Castaneda AR, Jonas RA, Mayer JE, et al. Cardiac Surgery of the Neonate and Infant. Philadelphia: W.B. Saunders; 1994:158, with permission. Copyright Elsevier.)
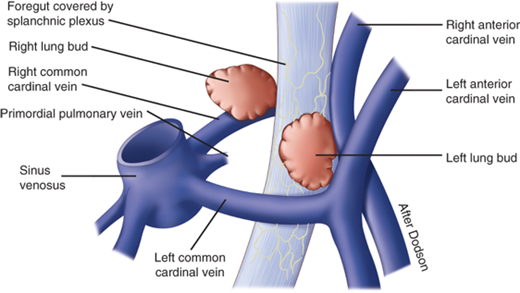
Darling and colleagues classified TAPVC according to the site or level of connection of the pulmonary veins to the systemic venous system73: type I (45%), anomalous connection at the supracardiac level; type II (25%), anomalous connection at the cardiac level; type III (25%), anomalous connection at the infracardiac level; and type IV (5%), anomalous connection at multiple levels.74 Within each category, further subdivisions can be implemented, depending on whether pulmonary venous obstruction exists. Obstruction to pulmonary venous drainage is a powerful predictor of adverse natural outcome and occurs most frequently with the infracardiac type, especially when the pattern of infracardiac connection prevents the ductus venosus from bypassing the liver.75
Because both pulmonary and systemic venous blood returns to the right atrium in all forms of TAPVC, a right-to-left intracardiac shunt must be present in order for the afflicted infant to survive. This invariably occurs via a nonrestrictive patent foramen ovale. Because of this obligatory mixing, cyanosis is usually present, and its degree depends on the ratio of pulmonary to systemic blood flow. Decreased pulmonary blood flow is a consequence of pulmonary venous obstruction, the presence of which is unlikely if the right ventricular pressure is less than 85% of systemic pressure.76
The child with TAPVC may present with severe cyanosis and respiratory distress, necessitating urgent surgical intervention if a severe degree of pulmonary venous obstruction is present. However, in cases where there is no obstructive component, the clinical picture is usually one of pulmonary overcirculation, hepatomegaly, tachycardia, and tachypnea with feeding. In a child with serious obstruction, arterial blood gas analysis reveals severe hypoxemia (partial pressure of oxygen [Po2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


