Congenital Amegakaryocytic Thrombocytopenia
David Czuchlewski, MD
Key Facts
Terminology
Autosomal recessive disease characterized by
Thrombocytopenia, usually presenting at birth
Absence of syndromic malformations or dysmorphism
Frequent evolution to multilineage bone marrow failure and pancytopenia
Etiology/Pathogenesis
Molecular analysis identifies MPL mutations in most CAMT patients
CAMT inherited loss of function MPL mutations must be differentiated from acquired activating MPL mutations associated with myeloproliferative neoplasms
Microscopic Pathology
Platelets are morphologically unremarkable
Megakaryocytes are absent or markedly reduced in number
Top Differential Diagnoses
Acquired thrombocytopenias are vastly more common than CAMT
Normal platelet size and absent megakaryocytes help to exclude inherited macro- and microthrombocytopenias
Fanconi anemia and dyskeratosis congenita can overlap with clinical presentation of CAMT and should be ruled out
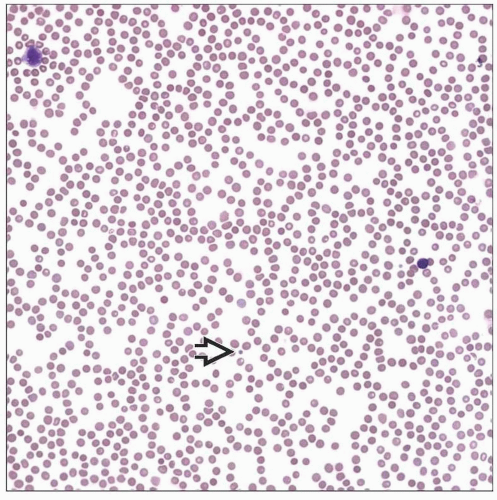 Patients with congenital amegakaryocytic thrombocytopenia present at birth or soon after with platelet counts of around 20,000/µL. Platelets that are present  are morphologically normal. are morphologically normal. |
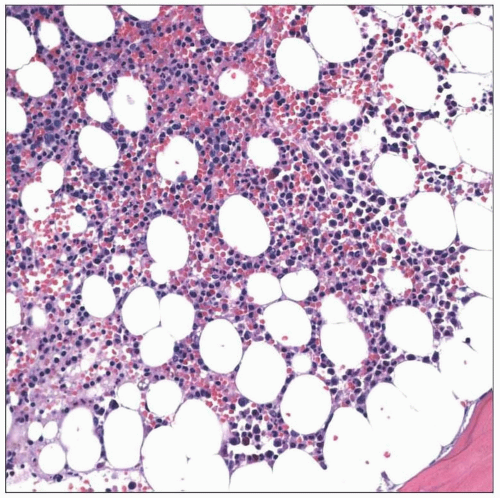 Congenital amegakaryocytic thrombocytopenia is characterized by absent or markedly reduced megakaryocytes. Other lineages are unaffected, and early in the disease course, cellularity is normal. |
TERMINOLOGY
Abbreviations
Congenital amegakaryocytic thrombocytopenia (CAMT)
Definitions
Autosomal recessive disease characterized by
Thrombocytopenia, usually presenting at birth
Absence of syndromic malformations or dysmorphism
Frequent evolution to multilineage bone marrow failure and pancytopenia
ETIOLOGY/PATHOGENESIS
Overview of Thrombopoietin (TPO) Signaling
TPO is produced at fairly constant rate
TPO is removed from circulation by binding to TPO receptors on platelets and megakaryocytes
TPO receptor is known as MPL (c-Mpl, CD110) and is encoded by gene MPL
As platelet count increases, more TPO is consumed by platelets bearing MPL and thus less is available to stimulate bone marrow production
Intact THPO and MPL expression is necessary for both megakaryopoiesis and proper maintenance of other bone marrow progenitor cells
In knockout mouse models, absence of functional MPL results in reduction of both platelets and stem cells to ˜ 10% of normal
MPL activation occurs via TPO binding and receptor dimerization
Further intracellular signaling relies upon JAK-STAT mechanism
MPL Mutations and CAMT
CAMT is caused by homozygous or compound heterozygous loss of function mutations in MPL
Clinical features are to some extent determined by location of mutation and consequent degree of alteration to the protein
Type I mutations are severe mutations that essentially abrogate MPL functionality
Often these are nonsense mutations
Type II mutations preserve some MPL function
These are often missense or splice site mutations
CAMT-associated MPL mutations must be differentiated from unrelated MPL variants associated with several clinical presentations
Activating MPL somatic mutations
In contrast to inherited loss of function mutations in CAMT, these acquired activating mutations increase MPL signaling and result in thrombocytosis
Acquired activating MPL mutations are seen in some myeloproliferative neoplasms
MPL polymorphisms not associated with CAMT
Single nucleotide polymorphism (SNP) G1238T (clinically termed MPL Baltimore) is associated with reduced MPL expression but paradoxical thrombocytosis
G1238T is most prevalent in African-Americans
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


