CNS Primitive Neuroectodermal Tumors
Fausto J. Rodriguez, MD
Key Facts
Terminology
Heterogeneous group of embryonal neoplasms composed of poorly differentiated cells that may express neuronal and glial markers
Clinical Issues
Predominantly tumors of children, but may present in adults
Cerebral hemispheres are most frequent location
Occasional cases in suprasellar region, brainstem, and spinal cord
PNETs as a category are highly aggressive neoplasms with propensity for CSF dissemination and metastasis outside CNS
Worse prognosis as group than medulloblastoma
Not all PNETs are equivalent
Microscopic Pathology
Hypercellularity, round to oval crowded nuclei with stippled chromatin
Elevated mitotic activity and frequent apoptotic bodies
Presence of severe anaplasia at increased frequency (approximately 50% of PNETs) compared with medulloblastoma
Relatively circumscribed, but may infiltrate adjacent brain tissue in some cases
Ancillary Tests
Synaptophysin expression most common
Also may express neurofilament protein, chromogranin-A, NeuN
GFAP expression variable
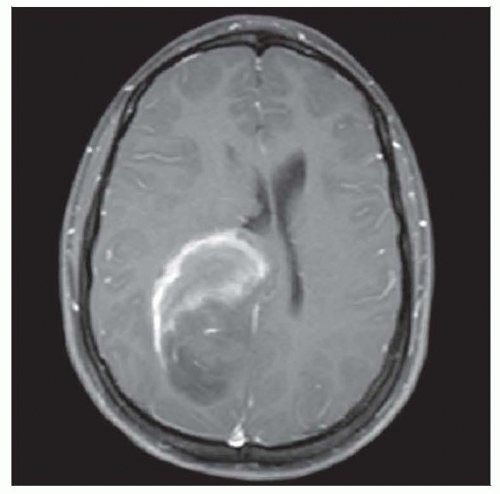 CNS-PNETs may be large, enhancing masses. This lesion has a rim pattern; others are solidly enhancing, and some have surprisingly little enhancement given the overt histological abnormality. |
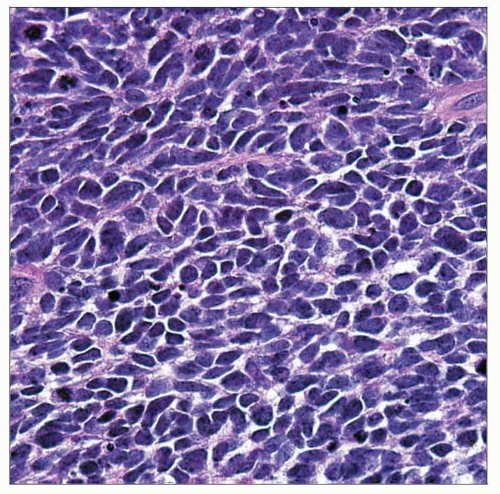 CNS-PNETs are highly cellular neoplasms whose diagnosis in undifferentiated cases rests largely on immunohistochemical findings (and even then can be difficult). |
TERMINOLOGY
Abbreviations
Central nervous system primitive neuroectodermal tumor (CNS-PNET)
Synonyms
Embryonal tumor with abundant neuropil and true rosettes (ETANTR)
Definitions
Heterogeneous group of embryonal neoplasms composed of poorly differentiated cells that may express neuronal and glial markers
To be distinguished from primitive neuroectodermal tumor (PNET)-like tissue in some malignant gliomas (e.g., glioblastoma)
Unrelated to peripheral PNET (Ewing sarcoma)
WHO grade IV
CLINICAL ISSUES
Epidemiology
Age
Predominantly in children (rarely in adults)
Site
Cerebral hemispheres most frequent
Near ventricular system in many cases
Occasionally suprasellar, brainstem, and spinal cord
Presentation
Symptoms secondary to mass effect, hydrocephalus
Treatment
Craniospinal irradiation and multimodality chemotherapy
Prognosis
Highly aggressive with a propensity for CSF dissemination
Overall worse prognosis than medulloblastoma
May vary by age (children vs. adults)
IMAGE FINDINGS
MR Findings
Relatively circumscribed compared to infiltrating gliomas
Variable contrast enhancement
Spinal MR required in most cases to exclude leptomeningeal dissemination
CT Findings
Hyperdensity secondary to high cellularity
Minority may calcify
MICROSCOPIC PATHOLOGY
Histologic Features
Hypercellularity, round to oval crowded nuclei with stippled chromatin
Elevated mitotic activity and frequent apoptotic bodies
Relatively circumscribed, but may infiltrate
Variable neuronal and glial differentiation
Immunohistochemistry for confirmation
Distinction from small cell malignant gliomas problematic, especially in cases with glial differentiation
Cerebral Neuroblastoma
Ganglioneuroblastoma
Neuroblastic differentiation but also containing mature ganglion cells
Significance of only a few neuroblasts unclear, but treatment similar to PNET
Medulloepithelioma
Rare subtype predominantly in very young children
Less frequent stereotypical locations include ciliary body, retina, and optic nerve
Supra- or infratentorial
Luminal mitoses
Ribbons with obvious epithelial features, but primitive pseudostratified cells and external, PAS positive, limiting membrane
Epithelioid ribbons have morphologic overlap with embryonic neural tube
Ependymoblastoma
Ill-defined category, existence recently challenged
Traditionally defined by presence of “ependymoblastic” or multilayered rosettes in an otherwise conventional PNET
Rosettes mitotically active
Many ependymoblastomas fall into other more specific PNET categories, usually either embryonal tumor with abundant neuropil and true rosettes (ETANTR) or medulloepithelioma
Embryonal Tumor with Abundant Neuropil and True Rosettes
Rare aggressive tumor, usually of 1st 2 years
Large, often well-circumscribed mass with little contrast enhancement
Highly cellular undifferentiated tissue, prominent neuropil and distinctive rosettes with sharply defined lumina
Neurocytes and small ganglion cells in neuropil areas
Genetically distinct with frequent amplification at chromosome 19q13.41 microRNA polycistron
Undifferentiated Small Cell Embryonal Tumors
Subset of tumors, particularly in children, may not demonstrate specific line of differentiation by morphology or immunohistochemistry
Frequent difficulty arises in categorizing such lesions as PNET, high-grade glioma, or undifferentiated primary CNS tumor
Nodular Subtype
Incompletely characterized
Similar in part to nodular/desmoplastic medulloblastoma
Synaptophysin (+) nodules but no perinodular reticulin



