Overview
Lipids perform several essential functions, including forming biological membranes, efficient storage of energy, and as components of several important structural and functional molecules. Lipid metabolism includes both the synthesis and degradation of fatty acids and/or more complex lipid molecules. The choice between synthesis and degradation represents an important regulatory step in human biology and reflects the level of food and, therefore, energy stores available to the body. Several processes are involved in this decision of producing fatty acids/lipids or, instead, directing their precursors to energy production via carbohydrate metabolic pathways. Separate but still dependent on this process, the production of cholesterol and several lipid-derived hormones and signaling molecules is essential for multiple functions in the human body. As in most metabolic pathways, deficiencies or problems can lead to serious disease states. As seen in other chapters, understanding of the exact mechanism of these problematic metabolic steps also allows treatment of these diseases.
Fatty Acid Metabolism
Although most fatty acids needed by humans are supplied in the diet, fatty acid synthesis plays a role in certain tissues to convert any excess of sugar molecules into the more efficient storage form of fatty acid/lipid molecules and/or to produce specialized lipid molecules. The link between sugar and fatty acid/lipid metabolism is seen at acetyl coenzyme A (CoA), the intermediate between glycolysis and the citric acid cycle. As will be seen below, acetyl-CoA is also the initial molecule of fatty acid metabolism. Therefore, acetyl-CoA is an important branch point of human metabolism and its use is highly controlled to reflect the body’s nutritional state and needs. As an illustration of this fact, the first and committing step of fatty acid synthesis is the addition of a carboxyl (CO2−) group, donated by HCO3−, to acetyl-CoA to produce malonyl-CoA.
Although the reaction is simple, the choice to commit the potential energy production from carbohydrates to produce lipid molecules reflects several important biological principles in human metabolism. First, the enzyme that catalyzes this reaction, acetyl–CoA carboxylase, is produced as smaller, inactive protein complexes. When the concentration of citrate is high (the first intermediate of the citric acid cycle and a sign of abundant sugar resources), these smaller complexes join to form active enzymatic polymers (Figure 7-1). Therefore, citrate increases fatty acid synthesis when the body has plentiful energy and needs to store this energy in an efficient fashion. If lipid stores are high, though, increased concentration of palmitoyl-CoA (the final product of fatty acid synthesis) decreases fatty acid synthesis by depolymerizing the active enzyme complex into the original smaller, inactive complexes. The balance between citrate and palmitoyl-CoA or, more simply, a measure of sugar versus fat levels (e.g., high sugar/fat indicates plentiful food supplies; low sugar/fat indicates low food and/or starvation conditions) controls the activity of this committing step and, therefore, utilizes available energy supplies most efficiently.
Figure 7-1.
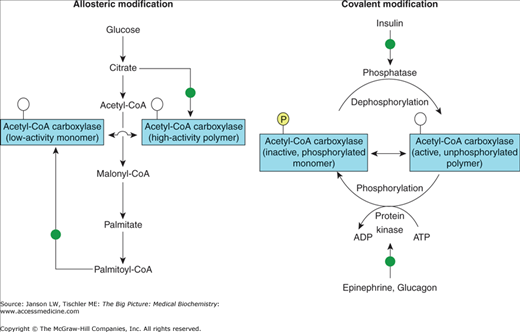
Regulation of the Acetyl-CoA Carboxylase and Production of Malonyl-CoA by Hormones. Palmitate and citrate regulate malonyl-CoA production allosterically by enhancing the formation of the low-activity monomer or the highly active polymer form of acetyl-CoA carboxylase, respectively. Both forms are unphosphorylated ( ). Insulin, epinephrine, and glucagon regulate acetyl-coA carboxylase activity by infl uencing the phosphorylation (inactivation,
). Insulin, epinephrine, and glucagon regulate acetyl-coA carboxylase activity by infl uencing the phosphorylation (inactivation,  ) or dephosphorylation (activation,
) or dephosphorylation (activation,  ) of the monomer. [Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
) of the monomer. [Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
There is a second, longer term, hormonal regulation that allows the human body even further control of energy sources. Glucagon, the hormone that promotes the breakdown of glycogen to glucose, and/or epinephrine, released in times of stress when energy requirements are higher, activate phosphorylation of acetyl-CoA carboxylase, causing inhibition of fatty acid synthesis (Figure 7-1). Alternatively, insulin, the hormone that promotes uptake and storage of sugars (Chapter 10), causes dephosphorylation, which increases production of malonyl-CoA and, therefore, fatty acid synthesis. Therefore, glucagon and epinephrine drive acetyl-CoA molecules away from lipid storage and toward the production of energy via the citric acid cycle. Additionally, food deprivation or chronic high-fat diet decreases the amount of the enzyme that produces malonyl-CoA.
Following the committing step of malonyl-CoA production, fatty acid synthesis is a relatively simple pathway that takes place on a multienzyme–protein complex in the cytoplasm. This enzyme complex, called fatty acid synthase, contains seven separate subunits, catalyzing linked enzymatic reactions, and an acyl carrier protein “arm,” which carries the growing fatty acid chain through to palmitate, the 16-carbon-long fatty acid with single carbon–carbon bonds. The overall reaction for production of one palmitate molecule, including the seven adenosine triphosphate (ATP) molecules to generate malonyl-CoA via acetyl-CoA carboxylase, is as follows:
Other fatty acids are derived from palmitate by specific enzymatic reactions. Production of fatty acids with an odd number of carbon atoms relies on the substitution of the three-carbon, propionyl-CoA, molecule for acetyl-CoA to initiate the fatty acid synthase reaction. The basic process of fatty acid production, which relies on the cofactor nicotinamide adenine dinucleotide phosphate (NADPH) to drive the reaction, is shown in Figure 7-2. Similar to the malonyl-CoA reaction, the quantity of the enzyme complex is positively regulated by higher sugar levels and negatively regulated by high fatty acid/fat levels in the diet.
Figure 7-2.
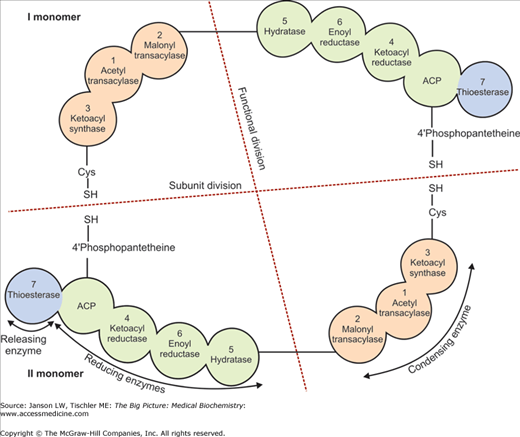
Fatty Acid Synthase. The human fatty acid synthase enzyme complex is composed of two identical monomers (I and II, “subunit division”), which are, themselves, split into condensing and reducing enzyme halves (“functional division”). The enzymes responsible for the growing fatty acid chain are lined up (although not in the linear order of reactions), starting at ketoacyl synthase. As one reaction concludes, the product is carried to the next enzyme by an “acyl carrier protein” (ACP), a section of the enzyme complex, which can flip back and forth between the successive enzyme reactions. Building blocks of acyl or malonyl subunits are attached to the ACP by acetyltransacylase or malonyltransacylase, respectively. Reducing enzymes creates the final carbon–carbon bond of the growing fatty acid chain. The new fatty acid product either returns to the start of the process or, once a 16-carbon, palmitate molecule is achieved, is released by thioesterase. The exact tertiary and quaternary structure of fatty acid synthase is still in question but all models agree with the successive enzymatic processes and the role of the ACP. [Reproduced with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
[Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.
Palmitate is used to produce longer fatty acid chains by the addition of more two-carbon units from malonyl-CoA as indicated by the equation below. This reaction mainly occurs in the endoplasmic reticulum by a four-step process including the enzyme fatty acid elongase with continued use of NADPH as a source of both energy and reducing power. Fatty acid elongation can also occur in the mitochondria (NADH dependent) and in selected tissues (see Sidebar) where particular fatty acid types are required.
Finally, the production of carbon–carbon double bonds in fatty acids takes place via a desaturase enzyme, utilizing NADH, in the endoplasmic reticulum.
[Adapted with permission from Naik P: Biochemistry, 3rd edition, Jaypee Brothers Medical Publishers (P) Ltd., 2009.]
Lipid Synthesis in Selected Tissues: New synthesis of fatty acids in the human body takes place in liver, adipose (fat) tissue, kidney, brain, and the mammary glands. The liver is intimately involved in maintaining the balance of lipids, including cholesterol (Chapter 11). Adipose tissue, commonly referred to as fat cells, plays a key role in the storage of lipids in the form of triacylglycerol molecules. Triacylglycerol can store 9 kilocalories of energy per gram (kcal/g) versus only 4 kcal/g for carbohydrates, making triacylglycerol the most efficient form of energy storage in the body. Fatty acid, triacylglycerol, and cholesterol syntheses are also found in kidneys, and increased levels of lipids are seen in various kidney diseases (Chapter 18). The brain is one of only a few tissues that use ketone bodies (reviewed below) as an energy source. Because the brain uses approximately 25% of the body’s total energy production, this alternative energy source to carbohydrates most certainly illustrates an essential adaptation for survival during periods of reduced food intake. The brain also synthesizes lipid molecules essential in the growth and maturation of the nervous system from infancy to adulthood. Lack of these synthetic functions leads to a number of neurological diseases. Mammary glands also respond during pregnancy by producing triacylglycerol containing medium-chain fatty acids found in human milk. These lipids, which make up 3%–5% of human breast milk, are essential for the development of the newborn. |
Essential Unsaturated Fatty Acids: Humans cannot produce desaturated bonds beyond the ninth and tenth carbons and, therefore, need to acquire fatty acids with more distal double bonds from dietary sources (Chapter 3). These essential, unsaturated fatty acids are required for production of molecules such as prostaglandins (important in inflammatory reactions, various aspects of pregnancy, the spread of some cancers, control of blood flow to the kidney, regulation of ion flow across membranes, the conduction of nerve impulses, and modulation of sleep), thromboxanes (required for blood clotting and possibly involved in constriction of blood vessels, e.g., Prinzmetal’s angina), and leukotrienes (important molecules in immune reactions including asthmatic and allergic reactions as well as certain cardiovascular and neuropsychiatric diseases). |
The process of the breakdown or degradation of fatty acids starts in the cytoplasm with the linkage of a fatty acid to CoA forming a fatty acyl-CoA molecule. Following this linkage, fatty acids less than 12 carbons in length cross the mitochondrial membranes without assistance. However, fatty acids longer than 12 carbons are transported into the mitochondria via a unique process involving the molecule carnitine. In the first of three steps, the fatty acyl-CoA links to carnitine by the enzyme carnitine palmitoyltransferase I (CPT I), located on the outer mitochondrial membrane (Figure 7-3). The new molecule moves across the mitochondrial inner membrane via a specific membrane protein called translocase. Inside the mitochondria, CPT II, located on the inside of the inner mitochondrial membrane (Figure 7-3), removes the carnitine molecule. This carnitine molecule moving out of the mitochondria exchanges with a new molecule of palmitoylcarnitine moving into the mitochondria. High concentration of malonyl-CoA, the molecule representing commitment to the synthesis of fatty acids, prevents the transport of fatty acids into mitochondria by inhibiting CPT I. Increased amounts of fatty acyl-CoA reverse this inhibition and stimulates fatty acid degradation.
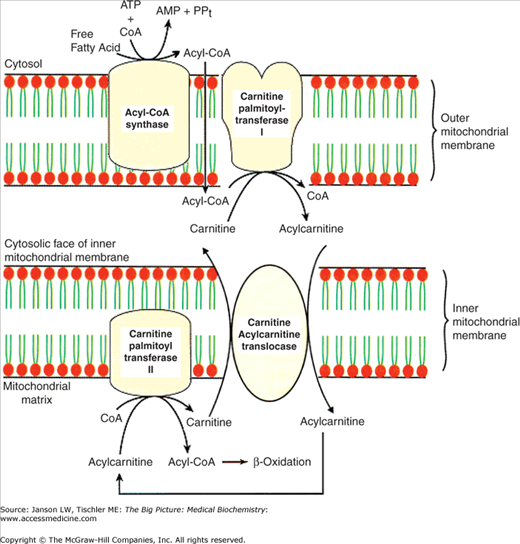
CPT Diseases: Not surprisingly, deficiencies in CPT I or II lead to severe disease states. CPT I deficiency is an autosomal recessive disorder that usually strikes in infancy or early childhood and causes low sugar, low ketone levels, and increased ammonia levels in association with fasting or illness (i.e., when the body attempts to use fatty acids as an energy source). Patients with CPT I deficiency can develop damaged and enlarged livers (partly due to the buildup of fatty acids), kidney problems, muscle breakdown, seizures, coma, and impaired growth. Without a diet of only medium-chain triglycerides (whose fatty acids can cross the mitochondrial membranes independent of carnitine) and the avoidance of fasting, death is common. Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|